实验室常用实验方法
公共平台 李敏敬
总RNA的提取(Trizol法提取)
在收集到生物材料之后,最好能即刻进行RNA制备工作。若需暂时储存,则应以液氮将生物材料急速冷冻后,储存于-80℃冷冻柜。在制备RNA时,将储存于冷冻柜的材料取出,立即以加入液氮研磨的方式打破细胞,不可以先行解冻,以避免RNase的作用。
1. 提取组织RNA时,每50~100mg组织用1ml Trizol试剂对组织进行裂解;提取细胞RNA时,先离心沉淀细胞,每5-10 ╳106个细胞加1ml Trizol后,反复用枪吹打或剧烈振荡以裂解细胞;
2. 将上述组织或细胞的Trizol裂解液转入EP管中,在室温(15℃~30℃)下放置5分钟;
3. 在上述EP管中,按照每1ml TRIZOL加0.2ml氯仿的量加入氯仿,盖上EP管盖子,在手中用力震荡15秒,在室温下(15℃~30℃)放置2~3分钟后,12000g(2℃~8℃)离心15分钟;
4. 取上层水相置于新EP管中,按照每1ml TRIZOL加0.5ml异丙醇的量加入异丙醇,在室温下(15℃~30℃)放置10分钟, 12000g(2℃~8℃)离心10分钟;
5. 弃上清,按照每1ml TRIZOL加1ml 75%乙醇进行洗涤,涡旋混合,7500g(2℃~8℃)离心5分钟,弃上清;
6. 让沉淀的RNA在室温下自然干燥;
7. 用Rnase-free water 溶解RNA沉淀。
PCR
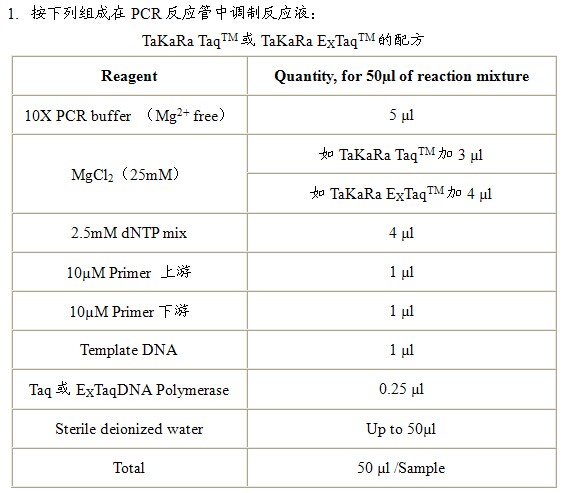
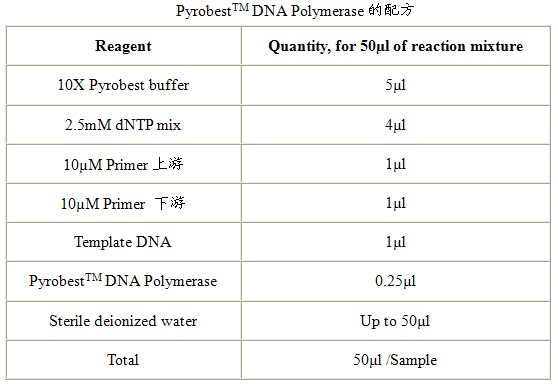
实验室常用DNA聚合酶有三种:TaKaRa TaqTM,TaKaRa EXTaqTM和PyrobestTM DNA Polymerase。TaKaRa TaqTM是一般的DNA聚合酶,保真性较差,但价钱便宜,一般用于基因表达的检测等。TaKaRa EXTaqTM是具有Proof reading活性的耐热性DNA聚合酶,具有一定的保真性,而且其扩增得到的PCR产物3’端附有一个“A”碱基,如果希望直接将产物克隆到T-vector可以用此酶。PyrobestTM DNA Polymerase也是具有Proof reading活性的耐热性DNA聚合酶,其特点是保真性极高,扩增得到的PCR产物为平滑末端。如果进行基因的扩增请使用此酶。



琼脂糖核酸电泳
1. 用蒸馏水将制胶模具和梳子冲洗干净,放在制胶平板上,封闭模具边缘,架好梳子;
2. 根据欲分离DNA片段大小用凝胶缓冲液配制适宜浓度的琼脂糖凝胶:准确称量琼脂糖干粉,加入到配胶用的三角烧瓶内,定量加入电泳缓冲液(一般20~30 ml);
3. 放入到微波炉内加热熔化。冷却片刻,加入一滴荧光染料,轻轻旋转以充分混匀凝胶溶液,倒入电泳槽中,待其凝固;
4. 室温下30~45分钟后,凝胶完全凝结,小心拔出梳子,将凝胶安放在电泳槽内;
5. 向电泳槽中倒入电泳缓冲液,其量以没过胶面1mm为宜,如样品孔内有气泡,应设法除去;
6. 在DNA样品中加入10×体积的载样缓冲液(loading buffer),混匀后,用枪将样品混合液缓慢加入被浸没的凝胶加样孔内;
7. 接通电源,红色为正极,黑色为负极,切记DNA样品由负极往正极泳动 (靠近加样孔的一端为负)。一般60~100V电压,电泳20~40min即可;
8. 根据指示剂泳动的位置,判断是否终止电泳;
9. 电泳完毕,关上电源,在凝胶成像仪上观察电泳带及其位置,并与核酸分子量标准Marker比较被扩增产物的大小。

胶回收纯化DNA
1. 琼脂糖电泳,将特异电泳带用刀切下放入到EP管中,称琼脂糖带的重量;
2. 按照每100mg加400µl的量加入binding buffer,放入到EP管振荡器中,45℃~55℃温育振荡,直到所有的琼脂糖都溶解(大概要5分钟);
3. 取出纯化柱,将上述溶解液转移至柱中,室温下放置2分钟,8,000rpm 离心1分钟,弃EP管中的液体,将纯化柱放回EP管中;
4. 加500µl的wash buffer至柱中,8,000rpm 离心1分钟。弃管中的溶液;
5. 重复操作4步的操作1次,最后将纯化柱放入EP管中10,000rpm离心30秒,除去痕量的wash buffer;
6. 将纯化柱放入一个新的EP管。加30~40µl H2O或者elution buffer至纯化柱膜的中央,在37℃或50℃下放置2分钟,10,000rpm离心1分钟洗脱DNA,将EP管中的DNA溶液放在-20℃保存。
7. 注:若想要不电泳而直接纯化DNA溶液,只需要在第2步中按100µl液量加400µl的binding buffer,其余的步骤不变。
大肠杆菌质粒DNA的提取(碱裂解法)
此方法适用于小量质粒DNA的提取,提取的质粒DNA可直接用于酶切、PCR扩增。
1. 取1.5ml细菌培养物于EP管中,4000rpm离心1分钟,弃上清液,使细菌沉淀尽量干燥;
2. 将细菌沉淀重悬于用冰预冷的100 µl溶液I (50 mmol/L葡萄糖,10 mmol/L EDTA pH 8.0,25 mmol/L Tris-HCl pH 8.0) 中,剧烈振荡;
3. 加入200 µl新配制的溶液II(0.2 mol/L NaOH,1%SDS(m/v)),盖紧EP管口,快速颠倒离心管5次,以混合混合物,确保离心管的整个内表面与溶液II接触,不要涡旋,置于冰浴中;
4. 加入150 µl预冷溶液III(每100 ml 的溶液III中含60 ml 5 mol/L 乙酸钾,11.5 ml冰乙酸,28.5 ml H2O),盖紧EP管口,反复颠倒数次,使溶液III在粘稠的细菌裂解物中分散均匀,之后将管置于冰上3~5分钟;
5. 在最大转速下离心5min,取上清液于另一新EP管;
6. 用两倍体积的乙醇室温沉淀双链DNA,振荡混合于室温放置2分钟,最大转速离心5分钟;
7. 小心吸去上清液,将离心管倒置于滤纸上,以使所有液体都流出,在将附于管壁的液滴除尽;
8. 加1ml 70%乙醇洗涤沉淀,振荡混合,用12,000g离心2分钟,弃上清,将开口的EP管置于室温使乙醇挥发,直至EP管中内没有可见的液体存在(5~10分钟),用适量的ddH2O溶解;
9. 用0.5µl的RNase 37℃温育5~10分钟;
10. 电泳鉴定。
乙醇沉淀DNA
1. 加入1/10体积的乙酸钠(3 mol∕L,PH=5.2)于DNA溶液中充分混匀,使其最终浓度为0.3 mol∕L;
2. 加入2倍体积用冰预冷的乙醇混合后,再次充分混匀,置于-20℃中15~30分钟;
3. 12,000 g离心10分钟,小心移出上清液,吸去管壁上所有的液滴;
4. 加入1/2离心管容量的70%乙醇,12000g离心2分钟,小心移出上清液,吸去管壁上所有的液滴;
5. 于室温下将开盖的EP管的置于实验桌上以使残留的液体挥发至干;
6. 加适量的ddH2O溶解DNA沉淀。
酶 切
1. 酶切前确定待切样品的浓度, 并选择合适的限制性内切酶和配套Buffer。
2. 在离心管中加入如下成分:
10×Buffer 1μl
待切样品 xμl
酶 0.5-1μl
 加水补足 10μl
加水补足 10μl
3. 混匀样品并短暂离心使样品沉于管底。
4. 将离心管置于37℃中温育1-3hr,若待切样品为PCR产物,则可将反应时间适当延长。
5. 用未酶切的质粒作为对照,琼脂糖电泳鉴定酶切结果。
注:当酶切样品用于回收而不是鉴定时,可按比例适当加大反应体积。双酶切可选用二者活性都较高的Buffer或者通用Buffer,但要注意不能有星反应。)
连 接
1. 连接前先电泳确定待连接载体与片段的浓度。
2. 在离心管中加入如下成分:
10×连接Buffer 1μl
待连接的样品(胶回收产物或PCR产物,载体与片段的mol比为1∶3-5)
连接酶 0.5-1μl
 加水补足 10μl
加水补足 10μl
3. 混匀样品并短暂离心使样品全部沉于管底。
4. 将离心管置于连接酶要求的温度孵育适当的时间(根据不同公司的酶的要求而定,一般为22℃ 1-3hr或16℃连接过夜)。
连接完的样品可直接用于转化,也可放4℃冰箱短期保存。
感受态细胞的制备
1. 挑取适当菌株的E.coli 单菌落接种于2ml SOB培养液中,37℃摇床过夜。
2. 取0.5-1ml过夜培养的菌液转种到50ml SOB中,18℃剧烈震荡,直到A600达到0.6。
3. 将培养物转移到50ml离心管中,4℃ 4,000rpm/min离心10min。同时在冰浴上配置TB溶液。
4. 弃上清,将离心管倒置于滤纸上,使培养液被吸干。
5. 取1ml刚配的TB溶液打散菌体沉淀,再加入15ml TB(1/3体积的起始培养液),冰浴10-15min,4℃ 4,000rpm/min离心10min。
6. 弃上清,沉淀重悬于4ml TB(1/12.5体积的起始培养液),冰浴10min。
7. 加入280μl DMSO,缓缓滴入并轻轻摇晃,使其充分混合均匀,冰浴10min。
8. 将菌液分装于EP管中,-80℃或液氮冻存。
9. 取两管感受态细胞分别加入1μl无菌ddH2O(阴性对照)和1μl纯质粒(阳性对照)进行转化(见后),以检测感受态的质量。阴性对照平板上应该无菌落生长,阳性对照平板上菌落数目的多少显示感受态效率的高低。
SOB的配制:
蛋白胨 20g
酵母提取物 5g
NaCl 0.58g
KCl 0.186g
100×Mg++溶液 10ml
 溶解并加水定容至1L,121℃×20min高压蒸汽灭菌
溶解并加水定容至1L,121℃×20min高压蒸汽灭菌
100×Mg++溶液:
MgCl2﹒6H2O
MgSO4﹒7H2O
溶解并加水定容至100ml,121℃×20min高压蒸汽灭菌
TB溶液的配制:
1M KCl 5ml
0.55M MnCl2 2ml
0.5M CaCl2 0.6ml
0.1M K-Pipes(pH 6.7) 2ml
ddH2O 10.4ml
 Total 20ml
Total 20ml
注:上述溶液均需高压蒸汽灭菌处理
0.1M K-Pipes(pH=6.7)的配制:
称取3.02g Pipes粉末溶于80ml dd H2O中,此时粉末不能完全溶解,用10N KOH或KOH固体调节PH值,只有当PH接近6.7时粉末才能完全溶解,此时当小心少量地加入KOH直至达到所需PH值。
转 化
1. 取100μl感受态细胞于冰浴上融化。
2. 加入1μl纯质粒或连接产物,轻轻吹打混匀,冰浴30 min。
3. 将菌液放入42℃水浴中热激90秒,立即放入冰浴中2 min。
4. 加入0.9ml SOC,于37℃恒温摇床上200rpm×1hr温育。
5. 将菌液4000rpm/min离心3min,留200μl上清将菌体打散,均匀涂布于含适当抗生素的琼脂平板表面,平板于37℃倒置培养过夜。
i. 注:新倒的平板可于37℃培养箱中预先放置数小时至过夜干燥。
ii. 当转化的是TA克隆连接产物时可在菌液中加入8μl 1M IPTG和40μl 20mg/ml X-gal以进行蓝白斑筛选。
重组子的筛选和鉴定
重组子可通过酶切进行鉴定,也可以利用扩增引物通过PCR进行鉴定,阳性重组子能切出所需要的片段或得到相应片段的PCR产物。
1. 用牙签挑取平板上的菌落接种于2ml含适当抗生素的LB培养基中,37℃摇床培养过夜。
2. 次日取菌液0.2-0.5ml,13,000rpm×3min离心,弃上清,加入20μl ddH2O和20μl 酚/氯仿,震荡混匀,13,000rpm×5min离心。
3. 取上清进行琼脂糖电泳,加入载体质粒DNA作为阴性对照,根据质粒大小初步筛选重组子,重组子的泳动速度应该慢于载体质粒。
4. 用碱法小量制备可能是重组子的质粒DNA。
5. 选取适当的酶,对重组子进行酶切分析,酶切体积均为10μl体系。酶切样品进行琼脂糖电泳鉴定是否有所需片段。
6. 酶切分析正确的重组子分成两份,一份进行测序反应,另外一份保种。
7. 若用PCR法鉴定,则在第2步时每个样本取0.5-1.0μl 菌液为模板进行PCR反应,每管反应体系最低可少至10μl,PCR产物电泳,能得到所需条带的样本进一步提取质粒酶切鉴定或送样品测序。
真核细胞的转染
该操作以Invitrogen公司的脂质体转染试剂LipofectAMINE 为例,其它转染试剂可参照各自的使用说明书进行。
1. 在6孔板中接种1-3×105细胞/孔,加入2ml完全培养基,置CO2孵箱中37℃培养过夜。
2. 待细胞长到50-80%单层时,在无菌离心管中配制如下溶液:
i. 溶液A:将1-2μg待转染的超纯DNA稀释到100μl无血清培养基中
ii. 溶液B:将2-25μl LipofectAMINE稀释到100μl无血清培养基中
3. 混合溶液A和B,轻轻混匀,室温放置15-45min。
4. 用2ml无血清培养基轻轻洗涤细胞,加入0.8ml无血清培养基/孔,将脂质体复合物滴加到孔中,轻轻摇晃混匀,置CO2孵箱中37℃孵育2-24hr。
5. 用完全培养基替换转染液,继续培养。
6. 24-72hr后检测蛋白质的表达,或传代并加入选择性抗生素以筛选稳定表达株。
转染细胞的稳定筛选
1.确定抗生素作用的最佳浓度:
不同的细胞株对各种抗生素有不同的敏感性,因此在筛选前要做预试验,确定抗生素对所选择细胞的最低作用浓度。
1) 提前24小时在96孔板或24孔板中接种细胞8孔,接种量以第二天长成25%单层为宜,置CO2孵箱中37℃培养过夜。
2) 将培养液换成含抗生素的培养基,抗生素浓度按梯度递增(0, 50, 100, 200, 400, 600, 800 和1000μg/ml)。
3) 培养10-14天,以绝大部分细胞死亡浓度为准,一般为400-800μg/ml,筛选稳定表达克隆时可比该浓度适当提高一个级别,维持时使用筛选浓度的一半。
2.转染按前面的步骤进行。
3.转染72小时后按1:10的比例将转染细胞在6孔板中传代,换为含预试验中确定的抗生素浓度的选择培养基。在6孔板内可见单个细胞,继续培养可见单个细胞分裂繁殖形成单个抗性集落,此时可用两种方法挑选单克隆。
1) 滤纸片法:用消毒的5x5mm滤纸片浸过胰酶,将滤纸片贴在单细胞集落上10-15秒,取出粘附有细胞的滤纸片放于24孔板中继续加压培养。细胞在24孔板中长满后转入25cm2培养瓶中,长满后再转入75cm2培养瓶中培养。
2) 有限稀释法:将细胞消化下来后做连续的10倍稀释(10-2—10-10),将每一稀释度的细胞滴加到96孔板中培养,7-10天后,选择单个克隆生长的孔再一次进行克隆。
4.ELISA或Western blot检测单克隆细胞中外源蛋白的表达情况。由于不同克隆的表达水平存在差异,因此可同时挑选多个克隆,选择表达量最高的克隆传代并保种。
重组蛋白质的表达、纯化、复性和定量
按Qiagen公司的操作手册进行,具体步骤如下。
一、重组蛋白质的诱导表达
1. 挑取转化有质粒的单菌落,接种于3ml 选择性LB液体培养基中,37 oC,250 rpm/min振摇培养过夜。
2. 次日将培养过夜的菌液500 μl再接种于10 ml(1:20)选择性LB液体培养基中,37 oC,250 rpm/min振摇培养至光密度(OD600=0.6)时,取1 ml样本作为诱导前标本,10000g离心1 min收集菌体沉淀,-20 oC冻存备用。
3. 加入1 mol/L IPTG于菌液中,使IPTG终浓度为1 mM,37 oC,250 rpm/min振摇培养4~5小时。取1 ml样本作为诱导后标本,同上法收集菌体沉淀,-20 oC冻存备用。
4. 将诱导前后菌体沉淀用20 ~ 40 μl PBS(pH= 8.0)重悬,加入等体积的2×SDS上样缓冲液,煮沸加热5 min,SDS聚丙烯酰胺凝胶(SDS-PAGE)电泳分离,考马斯亮蓝染色3小时后,脱色观察结果。
5. 选取诱导成功的细菌克隆,扩大诱导规模,收集菌体沉淀,于-20 oC保存,准备做下一步分析及纯化。
二、重组蛋白质的分离纯化
重组蛋白质的可溶性鉴定
1. 将按上法诱导培养后收集的菌体重悬于裂解液1 (Lysis buffer under native conditions )中,然后在-80 oC低温冰箱中放置10 min。
2. 冰中解冻。
3. 在冰浴上用超声破碎仪破菌6次,每次10 sec,间歇10 sec,电压200-300 V。
4. 10000g,4oC,离心20 min,取上清(为溶液A),-20 oC保存;另将沉淀用同样裂解液1溶解(为溶液B),同样-20 oC保存,供后继分析使用。
5. 将上述A、B溶液和诱导前后的细菌进行SDS-PAGE电泳,考马斯亮蓝染色,比较分析重组蛋白质的溶解性。如果诱导表达的蛋白质位于A溶液中,则为可溶性蛋白;如果是在B溶液中,则为非可溶蛋白。
重组蛋白质为非可溶性蛋白(变性条件下)的分离纯化
1. 将菌体沉淀溶于适量裂解液2(Lysis buffer under denaturing conditions)中,室温下搅拌和吹打沉淀,避免泡沫生成。
2. 10000g,4 oC,离心30 min,收集上清液。
3. 将Ni-NTA Agarose充填柱子,并连接于Pharmarcia低压液相层析系统,用5倍柱体积的裂解液2平衡Ni-NTA Agarose,调节A280值至零线。
4. 将适量上清液上样到Ni-NTA Agarose柱子中,并用lysis buffer冲洗至A280值低于0.01。
5. 分别用5~10倍柱体积的清洗液1 和清洗液2(Wash buffer 1 and 2)清洗柱子,直至A280值低于0.01。
6. 用洗脱液(Elution buffer)洗脱重组蛋白质,在A280值监测下,收集出现峰线后含有重组蛋白的所有洗脱液。
三、重组蛋白质的复性、冻干和定量
纯化后的蛋白用梯度降低的尿素溶液缓慢透析,最终用0.01×PBS透析,透析后的蛋白质溶液经冻干成粉状。以牛血清白蛋白(BSA)为标准,采用BIO-RAD公司蛋白质定量试剂(protein assay)比色测定蛋白质的含量。
肿瘤细胞体外传代培养及保种
一. 细胞复苏与培养
将液氮或-80oC保存的肿瘤细胞于37oC 水浴, 快速溶化(慢冻快融) 用8.0ml培养基混匀已融化的肿瘤细胞悬液, 于1500转/分, 离心3分钟。弃上清, 再吸取8.0ml培养基质混匀细胞沉淀, 再1500转/分, 离心3分钟, 弃上清, 细胞沉淀用1.0ml培养基混匀, 备用。 另取一个75cm2方瓶, 加入14.0ml培养基质, 将上述制备的含肿瘤细胞悬液(1.0ml)加入此方瓶中, 于37oC,5%CO2孵育箱中培养。
若此肿瘤细胞悬浮生长, 大约3-4 天细胞基质会变黄, 5.0ml细胞悬液可传代一个方瓶培养, 可用3-4个方瓶培养, 一个方瓶中呈对数生长的肿瘤细胞可达1—1.5×107 个, 根据试验所需, 可决定传代的次数。 若此肿瘤细胞呈贴壁生长, 经过3—4天, 肿瘤细胞生长至80%—95% 单层时, 弃上清, 用0.5mM 的EDTA(难消化的肿瘤细胞用0.25%胰酶1.0ml), 处理肿瘤细胞大约 3—5分钟, 用倒置显微镜观察,当90% 的肿瘤细胞变圆时, 即可用弯管吹打并将消化的细胞转移到15ml离心管中, 于1500转/分下,离心3分钟, 弃上清, 加少许培养基混匀, 可传代3个75cm2方瓶扩大培养。
二. 细胞冻存
将对数生长的肿瘤细胞用1个75cm2 方瓶按上述方法收集, 于1500转/分 离心3分钟,弃上清, 用保种液(含10% DMSO的小牛血清)3.0ml混匀, 分别加入到2—3只保种管中, 写上肿瘤细胞名称, 时间, 保种者姓名, 放-80o C 保存, 次日将它们转移到液氮中保存(注: -80o C下可保存细胞半年至一年, 液氮可保存细胞5—10年, 甚至更长的时间)。以上所有物品均需经过高温灭菌( 121oC, 30分钟),培养基质则经过过滤(0.22uM)除菌, 所有操作均必须遵守无菌操作技术, 避免细菌、真菌、病原体、衣原体等污染。
肿瘤动物模型的建立
将对数生长的肿瘤细胞收集, 用无血清基质10.0ml, 于1500转/分, 离心3min, 连续洗3次, 最后用无血清基质混匀, 用血球计数器计算肿瘤细胞数量(平均5个中方格的细胞计数×104 即是肿瘤数/毫升)。 计算完肿瘤细胞总数, 再将肿瘤细胞密度调至4×106 个/ml, 每只小鼠腋下接种50ul(即2×105个肿瘤细胞), 2周左右可扪及肿瘤小节结。一般选取6—8周的小鼠,不同的肿瘤细胞接种的数量和动物不一样, 比如LL/2, B16, Hepa 可接种C57和BALB/C 小鼠, NS-1, EL-4, C26, Meth A可接种BALB/C 小鼠, H22接种昆明鼠。从肿瘤接种后可扪及小结节开始, 每3天用游标卡尺量肿瘤纵横大小(单位: mm), 至少连续一个月时间。注意要设计不同的实验组和对照组, 每组动物数一般为5—10只, 一般接种肿瘤6—8周后,小鼠的肿瘤可生长至直径为15—20mm (即小鼠会濒临死亡)时, 可眼球取血,分离血清并保存血清, 处死小鼠, 取肿瘤组织照相, 取部分肿瘤组织作冰冻组织切片(或-80℃ 保存), 作相应的免疫组织化学染色, 取部分肿瘤组织用3%中性的甲醛固定, 石腊包埋, 作常规HE染色.
小鼠尾静脉注射方法
在靠近实验台边缘处, 用大培养皿扣住小鼠, 左手抓住小鼠尾巴, 用酒精棉球擦尾巴, 可见到两侧静脉; 注射前应确认针管内无气泡, 注射时由尾尖开始, 顺向刺入。失败后再逐渐移向根部重刺, 若准确刺入静脉内, 推进时无阻力, 一般可注入0.1—0.5ml。
肿瘤蛋白疫苗预防性动物实验
一般肿瘤蛋白疫苗首次免疫剂量10ug/只小鼠, 与相应佐剂混匀, 在背部皮下注射, 第2次免疫间隔2周, 同样加佐剂在皮下注射; 第3次免疫间隔2周, 同样加佐剂在皮下注射;第3次免疫后2周, 用ELISA检测其血清效价,当效价达到要求时;在接种肿瘤细胞前3天,于腹腔或尾静脉加强注射20ug /只。
人脐静脉内皮细胞(HUVEC)培养
1. 将15-20cm长的新生儿脐带放入无菌的PBS溶液中储存。
(注:4℃下最多贮存24小时,室温下不超过6小时,否则废弃)
2. 用一个钝头的针头扎入脐带静脉管中,用无菌的PBS溶液冲洗3-5次,将污血冲洗干净为止。
3. 用手术钳夹紧脐带下端,加入15ml 的胶原酶(1mg/ml)室温下消化15-20分钟,并不时上下摇动脐带。
4. 消化完后,将下端手术钳松开,消化液流入一个50 ml无菌离心管中,用无菌的PBS溶液冲洗脐带2-3次。
5. 将收集液离心(2000转/分)3分钟。
6. 倒去上清,加入10ml M199培养基(加入10U/ml的bFGF),用弯管吹散细胞,将所有液体转入一个用明胶包被好的培养瓶中,37℃培养。
(注:每个培养瓶中加入3-4ml无菌的1%的明胶溶液,摇匀使得明胶溶液完全铺满瓶底,放入37℃孵育,最少2小时,用前将明胶溶液倒了即可,明胶包被有利于细胞贴壁。)
7. 培养24小时后,倒掉培养基,并用无菌的PBS溶液清洗2-3次,洗掉红细胞和死细胞,加入10 ml新鲜的 M199培养基。
8. 以后每2天换一次培养基(每次换掉2/3的培养基)。
9. 一般培养5-7天,细胞可长满至80-90%单层,这时可以传代。
10. 倒掉培养基,用无菌的PBS溶液清洗2-3次,加入2-3ml消化液(0.25%胰酶+0.1%EDTA)消化细胞,在显微镜下观察,一旦细胞变圆,即加入2-3倍的有血清的DMEM培养基终止反应。
11. 用弯管将细胞吹打下来,并将所消化的细胞转移到一个50 ml无菌离心管中,2000转/分,离心3分钟。
12. 倒掉上清,加入10ml新鲜培养基,一般一瓶细胞可传代3-4瓶。以后照此传代培养。
13. 一般传代2-3代(培养了20天左右)用于做各种实验效果最好。
实验动物免疫方案
1、抗原: 蛋白质、多肽、细胞器、细胞、组织等。
2、免疫方式:皮下注射、腹腔注射、静脉注射、肌肉注射等。
3、不同动物免疫所需抗原量(以蛋白免疫为例)
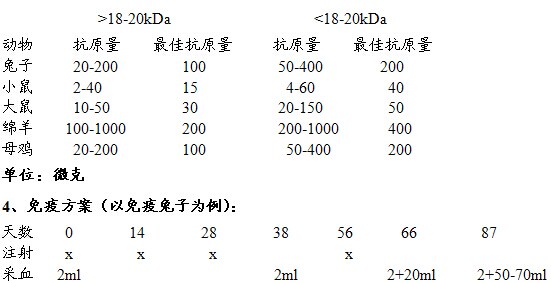
5、注意:
² 第一次免疫用完全佐剂与免疫原混合,以后加强免疫用不完全佐剂与免疫原混合,采取多点,时间间隔式免疫法。
² 如果用细胞免疫兔子,那么每次免疫所需细胞量为2-3 x 107 cells。
² 在连续免疫完三次后,需要少量取血进行ELISA检测,检测所免疫动物的抗体滴度,一般滴度能达到1:10,000-50,000。在处死所免疫的动物前一周应加强一次免疫。
² 如果用小鼠免疫,可以尾静脉小量采血(大约50µl),最后取眼球大量采血(大约500-1000µl);如果用兔子免疫,可以耳缘静脉小量取血(大约2mL),最后心脏大量取血(50-100mL)
² 按血清制备的标准方法将血清分离,并且分装成小份,储藏在-80oC。
血清制备
1. 取血后,37oC下,让血液凝固1到2小时(不加抗凝剂);
2. 4oC冰箱过夜(让血块固缩);
3. 当血清自然析出后, 4oC,3000转/分,离心10分钟,分离血清,弃去不溶物;
4. 将血清移至一干净试管,并分装成小份,储藏在-80oC。
ELISA
一、包被抗原
1. 用50mM的碳酸盐包被缓冲液(pH 9.6)溶解抗原,使抗原浓度为10-20 μg/ml,加100 μl/孔到96孔酶标板,4 oC放置过夜。
2. 第二天弃去包被液后,用PBST洗涤3次,每孔加入150 μl 1% BSA 37 oC封闭1小时。
3. PBST洗涤3次后,每孔加入100 μl不同倍比稀释度的血清,并加入对照样品,37 oC孵育2小时。
4. PBST洗涤5次后,加入100μl稀释后的HRP标记的二抗,37 oC孵育1小时。
5. PBST洗涤5次后,显色剂显色20 min后,酶标仪上读取A405吸收值。
二、包被细胞
1. 在96孔培养板上接种细胞数为1 x 104 cells/well,37℃过夜培养。
2. 第二天用PBS洗涤培养板2-3次。
3. 加入125 µl/well 10% Formalin(1:10稀释), 室温下固定15 min。
4. 用ddH2O洗涤培养板3次,并晾干,储藏在2-8oC备用。
5. 用PBST洗涤3次,每孔加入150 μl 1% BSA 37 oC封闭1小时。
6. PBST洗涤3次后,每孔加入100 μl不同倍比稀释度的血清,并加入对照样品,37 oC孵育2小时。
7. PBST洗涤5次后,加入100 μl稀释后的HRP标记的二抗,37 oC孵育1小时。
8. PBST洗涤5次后,显色剂显色20 min后,酶标仪上读取A405吸收值。

血清学筛选克隆新抗原/新基因
一、E.coli/ phage 裂解液预吸附血清
1. 将E.coli /phage lysate以1:10-20稀释在TBST溶液中。
2. 将4张82mm的nitrocellulose membranes(NC)浸入稀释后的E.coli/ phage lysate中,室温下水平摇动30分钟,取出NC并使膜沥干。
3. 用50ml TBST溶液洗膜3次,每次10分钟。
4. 用滤纸轻轻吸去膜上的液体。
5. 将膜放入50ml封闭液中,室温下水平摇动最少30分钟。
6. 将膜从封闭液中取出,用50ml TBST溶液洗膜3次,每次10分钟。
7. 将血清按1:5稀释在TBST溶液中,将一张膜放入溶液中,37℃下轻轻水平摇动10分钟。
8. 从血清稀释液中取出膜并丢弃,加入另外一张新膜,37℃下轻轻水平摇动10分钟。
9. 重复步骤8,直至所有4张膜都处理完。
10. 除去最后一张膜,收集血清(primary antibody),分装成小份储存于-80℃冰箱中待用。
注意:
² 该步处理过程是为了去除血清中能与细菌和噬菌体裂解蛋白进行免疫反应的抗体,这样可以减少假阳性率;
² 一抗不能反复冻融,化冻后不要再次冰冻,可放于4℃作短暂保存;
² 可以是病人血清,也可以是免疫血清,如果是病人血清,则需要至少10个病人血清进行混合;
二、噬菌体筛选
1. 准备NZY agar plates(至少用前24小时倒好),用前在37℃培养箱中烘烤1-2小时以去除水滴。
2. 将过夜培养的XL1-blue MRF’细菌2000转/分,离心10分钟,将细菌溶解在10mM MgSO4中,调整细菌浓度为OD600=0.5。
3. 融化NZY top,并将NZY top放在50℃水浴中。
4. 将适量的XL1-blue MRF’细菌溶液与一定稀释度的phage文库混合,37℃下共同作用15分钟。
² 直径90mm平板:200µl XL1-blue 细菌+适量的phage文库
² 直径150mm平板:600µl XL1-blue 细菌+适量的phag文库
(噬菌斑数量一般保持在3000pfu/90mm; 12000pfu/150mm)
5. 将步骤4中的混合液与NZY top溶液混合(200µl混合液+3-4mlNZY top溶液;600µl混合液+8-10 ml NZY top溶液),倒入到NZY agar plates中,室温下放10分钟左右,然后倒置放于37℃下培养。
6. 当噬菌斑刚好可看到时(大约5-8小时),从培养箱中拿出平板。
7. 将NC放入10mM IPTG溶液中完全浸湿,在空中使膜沥干,并做好3个不对称的标记。
8. 将IPTG处理好的NC贴在平板上,不留气泡,然后倒置放于37℃下培养。
9. 过夜培养后,第二天早上取出平板,用镊子将膜轻轻掀起,注意不要将培养基粘在膜上。
10. 将膜放于50ml TBST溶液中,水平脱色摇床上震荡洗膜3次,每次10分钟。
11. 将膜放入50ml封闭液中,水平摇动,封闭4-6小时。
12. 在封闭液中加入适当滴度的一抗,水平摇动处理过夜。
13. 将膜放于50ml TBST溶液中,洗膜3次,每次10分钟。
14. 在封闭液中加入适当滴度的二抗(各个公司的二抗使用滴度不同),室温水平摇动1-2小时。
15. 将膜放于50ml TBST溶液中,洗膜3次,每次10分钟,最后用50ml TBS溶液洗膜15-20分钟,取出膜空气中沥干。
16. 将膜放入BCIP-NBT显色液中避光显色,水平摇动直到阳性斑点可见为止。
17. 从显色液中取出膜放在TBS溶液中,空气中使膜干燥。
18. 根据所做的标记,将膜与平板对齐,将平板上对应的阳性克隆区域的培养基挖出放入500µl SM buffer中,并加入25 ml chloroform,4℃贮存(最多可贮存6月)。
19. 第一轮筛选得到阳性克隆需要进行第二轮筛选以去除假阳性并获得阳性单克隆噬菌体。过程如第一轮筛选,只不过用直径90mm平板;具体过程见步骤4,这时所加入的噬菌体溶液是第一轮筛选得到的噬菌体上清(见步骤18),在用前要滴定好噬菌体的滴度,噬菌斑的数量以可区分出单个克隆,同时密度不能太稀为标准(一般100-200 pfu/90mm)。
20. 按第一轮相似的过程进行实验,最后显色,确定真正的阳性克隆,并将阳性单克隆所在的培养基挖出放入500µl SM buffer中,并加入25 ml chloroform,4℃贮存(最多可贮存6月)。
注意:
² 封闭液一般可用:5%的脱脂牛奶或1%的BSA溶解在TBST溶液中。
² 第一轮筛选用150mm的平板;第二轮筛选用 90mm的平板,一般需要筛选至少1 x 106 pfu。
² 认真做好三个不对称的标记,特别在第二轮挑选阳性克隆时要仔细将膜与平板吻合好,不能挑错。
三、单克隆剪切
1. 取第二轮筛选得到的阳性克隆贮存液上清。
2. 将过夜培养的XL1-blue MRF’细菌2000转/分离心10分钟,将细菌溶解在10mM MgSO4中,调整细菌溶度到OD600=1.0。
3. 在一个EP管中加入:200µl XL1-blue MRF’细菌+250ul phage stock(步骤1)+1 ul ExAssist helper phage。
4. 将以上三种样品混合,37℃下共同保温15分钟。
5. 将样品混合物加入到3 ml LB培养基中,37℃震荡培养3-4小时。
6. 将试管放于65-70℃水浴20分钟,3000转/分,离心15分钟。
7. 将上清转入新的离心管中,已发生剪切的phage particles在上清中(上清可在4℃下储存1-2月)。
8. 将100ul phage 上清+200ul SOLR cells(OD600=1.0)混合,37℃保温15分钟。
9. 取步骤8中溶液5-10 ul涂于LB-amp agar plates(amp=50ug/ml),过夜培养。
10. 第二天细菌长出,随机挑取单克隆接种到LB-amp培养基中培养过夜。
11. 过夜培养细菌分为三部分:(1)提取质粒做双酶切,鉴定外源基因的大小;(2)送样品进行DNA测序(3)加入30-40%的甘油进行保种,分装成小份储存于-80℃备用。
附录:
1、SM buffer (1L):
(5.8 g NaCl+2.0 g magnesium sulfate+50 ml 1M Tris (pH=7,5)+0.1 g gelatin)
2、AP-buffer:
(100 mM Tris HCI (pH 9.5) ; 100 mM NaCl ; 5 mM MgCl2)
3、10xTBS(1L):
(0.1 M Tris-HCl (pH 8.0);1.5 M NaCl)
4、LB Broth(1L):
(10 g NaCl+10 g of tryptone+5 g of yeast extract)
ELISPOT
1. PBS溶解抗原为30 μg/ml,加100 μl/孔于PVDF膜铺底的96孔灭菌板过夜;
2. 第二天吸去包被液后,加5%FCS的PRMI 1640培养基100 μl封闭1小时,37 oC;
3. 准备脾细胞悬液(用氯化铵去除红细胞,制备成单个脾淋巴细胞悬液);
4. 从1 × 106/孔开始,按1:3的稀释度开始逐孔稀释做不同浓度梯度,并做3个复孔,37 oC静置培养5小时;
5. PBS洗3~5次,生物素化的抗鼠IgG二抗孵育30 min;
6. PBS洗3~5次,链亲和素标记的碱性磷酸酶孵育30 min;
7. PBS洗3~5次,用底物BCIP/NBT显色,显微镜下观察显色反应,显色后及时终止反应;
8. 计数每孔中的斑点数目,计算每106个脾细胞中抗体分泌细胞数量。
藻酸盐包裹实验
1. 将藻酸钠溶于无菌生理盐水,终浓度为1.5%;
2. 收集培养的肿瘤细胞,用无血清的培养基洗涤1次,将细胞沉淀重悬于1.5%藻酸钠溶液中;
3. 将上述肿瘤细胞悬浮液用1 ml加样枪缓慢滴入磁力搅拌的250 mM CaCl2溶液中,形成乳白色的藻酸盐小珠。继续静置于250 mM CaCl2中30 min即可使用。以上操作步骤均在无菌条件下进行;
4. 将第4次免疫后7天的小鼠用苯巴比妥钠0.1ml (按100 mg/kg计) 腹腔注射麻醉,小鼠麻醉后置于解剖台上,切开背部皮肤,皮下植入4粒藻酸盐包裹颗粒,缝合皮肤,外敷手术胶膜;
5. 天以后,小鼠经尾静脉注入100 μl (按100 mg/kg计) FITC标记的葡聚糖(FITC-Dextran);
6. 20 min后处死动物,取出藻酸盐颗粒,常温下加入1 ml的生理盐水,剪碎研磨颗粒,再加入1 ml的生理盐水,混匀标本,放置1小时,1500 rpm离心5 min。取上清液用荧光酶标仪测定;
7. 用不同浓度的FITC-Dextran制备标准曲线。
重组腺病毒构建,扩增及纯化基本技术操作
(细菌内同源重组AdEasy System)

一 目的基因的克隆(以pshuttle-CMV质粒为例)
1. 选择适当酶切位点,进行酶切连接,将目的片段插入pshuttl-CMV质粒多克隆位点。
2. 重组质粒鉴定: 酶切鉴定或测序。
3. 重组质粒扩增,纯化并准备适量含目的基因的穿梭质粒。
4. 用PmeI单酶切线性化重组穿梭质粒,电泳鉴定质粒完全被切开。
5. 胶回收线性化质粒,以备共转化使用。
二 共转化
1 大肠杆菌BJ5183电转感受态制备
² 从新鲜琼脂板上挑取单个BJ5183细菌,接种于LB培养基中,37℃振摇过夜。
² 接种25ml过夜培养物于500ml LB培养基,37℃振摇,至OD600 达到0.4。
² 迅速将培养物置于冰浴中30min,至充分冷却。
² 将菌液转移至预冷的离心管中,4℃下以2500r/min离心15 min,弃培养液,回收细胞。
² 以10ml预冷的10%甘油洗涤沉淀,低温离心,共两次。
² 加约1ml(适量)预冷的10%甘油重悬沉淀,稀释悬液100倍,测量OD600,至稀释浓度为2-3×1010个细胞/ ml (1.0 OD600约2.5×1010个细胞/ml)。分装,-80℃或液氮保存待用。
2 病毒骨架质粒转化大肠杆菌,扩增,纯化。
3 将1-5µl (约1µg) 线性化的穿梭质粒及1µl(约100ng/µl)病毒骨架质粒(如pAdEasy-1)加入至含约40µl BJ5183电转感受态的EP管中,混匀,冰上冷却。
4 将混合物加入电转杯,电击(1250-1500V/mm,5ms)。
5 电击结束取出样品,加入1ml SOC或LB培养液,37℃低速振荡40min。
6 取适当体积的电击转化细胞液涂于数个卡那霉素抗性平板(25-50mg/ml),37℃培养16-20hr。
7 次日挑取平板上长出的菌落(选择最小的菌落),接种于3ml含25-50mg/ml的LB培养基,37℃培养10-15hr。
8 碱裂法提取质粒,0.8%琼脂糖凝胶电泳筛选,大质粒为可能阳性克隆,进一步酶切鉴定。以PacI单酶切,0.8%琼脂糖凝胶电泳如显示出一条大片段(约30kb),及一条小片段(约3.0或4.5kb)(同时可进行其它酶切鉴定),则基本确定为阳性克隆。
9 取1-5µl 阳性质粒转化至DH5α大肠杆菌细胞(BJ5183为recA+,质粒DNA易发生突变,DH5α或JM109,XL1-blue菌株为重组缺陷性菌株,可稳定扩增已鉴定的重组质粒),扩增细菌并纯化质粒。
三 重组病毒质粒转导293细胞
1 293细胞培养:转导24小时前,以方瓶为例,接种2×106 293细胞于25cm2方瓶,使生长密度约50-70%。(也可以使用6孔或96孔板)
2 以PacI单酶切重组病毒质粒(转导25cm2方瓶约需4µgDNA),完全线性化后,乙醇沉淀,再以20 µl ddH2O溶解。
3 脂质体包裹质粒(以Lipofectamine为例):每4µg PacI约需20µl Lipofectamine包裹。质粒及脂质体分别稀释于500µl无血清培养基,再混合,置于室温下15-30min。
4 以无血清培养基轻轻洗涤培养瓶,另加2.5ml无血清培养基,37℃放置10min。
5 将Lipofectamine-DNA混合物加入培养瓶,37℃孵箱放置4hr。
6 4hr后,弃Lipofectamine-DNA混合液,另加入6ml DMEM完全培养基(含10% FCS)。如有大量细胞漂浮,可不弃Lipofectamine-DNA液,加入6ml DMEM完全培养基,37℃孵育过夜,再换液。
7 培养过程中观察细胞生长情况。约2周后可观察到细胞病变(CPE)出现(如用pAdTrack-CMV质粒,由于含GFP,可观察到绿色荧光)。
四 重组病毒鉴定
1 转导10-14d后,收集细胞沉淀,加入2ml灭菌的PBS混悬,冻融细胞,离心后收集上清保存于-80℃。
2 取30-50% 步骤1中上清,感染50-70%饱和度的25cm2方瓶中293细胞。2-3d后出现明显细胞病变。
3 感染后3-5d,当1/3-1/2细胞漂浮时收集病毒。按步骤1收集细胞并准备病毒上清。通过Western blot和/或PCR鉴定重组腺病毒产生。
4 PCR鉴定重组病毒。取5µl病毒上清加入10µl蛋白酶K,55℃孵育1hr,再煮沸5min,离心后取1-2µl作PCR。
五 重组病毒扩增,纯化
1 75cm2方瓶中接种293细胞,至密度达到90%,加入适量病毒上清感染细胞。3-4d后,细胞几乎变圆,且有一半细胞漂浮,则收集所有细胞。约500g转速离心,弃上清。
2 以灭菌PBS重悬沉淀,反复冻融4次。4℃下7000g离心5min.病毒纯化至少需要30瓶75cm2方瓶细胞。
3 CsCl连续梯度离心纯化:50ml离心管中称量4.4g CsCl,加入8ml病毒裂解上清液,混匀,体积约为10ml。转移至12ml超速离心管(用于SW41转头)中,覆盖约2ml矿物油。平衡后,10℃下32000 rpm离心18-24hr,用注射器抽吸离心出的病毒带。
(也可CsCl不连续梯度离心:20ml超速离心管中缓慢加入8ml CsCl 1.4 (53 g + 87 mL 10 mM Tris-HCl,pH=7.9),上面小心加入6 mL CsCl 1.2 (26.8 g + 92 mL 10 mM Tris-HCl,pH=7.9),再小心加入病毒上清液至体积达到20ml。平衡后,4℃下 23000rpm离心90min (SW28转头),用注射器抽吸下层蓝白色病毒带)
4 病毒透析去盐:配置透析液(10 mM Tris pH 8.0, 2 mM MgCl2, 5% sucrose),灭菌处理。4℃透析,更换3次透析液,可基本去除CsCl,病毒保存于-80℃。
六 病毒滴度测定(TCID50)
1 细胞准备:96孔板中接种100µl 293细胞,每孔细胞数约104个,以2% DMEM培养。
2 稀释病毒液准备:以2% DMEM将病毒液稀释成8个较高浓度(如10-3-10-10),每个浓度重复10个,每孔加入病毒稀释液100µl。另留两排不加病毒液作为阴性对照。37℃下,孵箱培养10天。
3 10d后观察细胞,记数每排出现CPE的孔数,计算细胞病变率。(如某一浓度各孔细胞全部病变,比率为1,如无细胞病变,则比率为0)。
4 计算T =10×101 + d (S - 0.5) /ml
d = Log 10 稀释度(如为10倍稀释℃,d=1)
S = 各浓℃细胞病变比率之和
实验室重组腺病毒常用质粒:病毒骨架质粒:pAdEasy-1, pAdEasy-2
穿梭质粒:p-Shuttle, p-Shuttle-CMV, pAdTrack, pAdTrack-CMV

组织病理技术
组织处理:
1. 取新鲜组织厚度不超过5mm,10% 中性福尔马林固定,大于24小时。
2. 固定后冲水12-24小时,
3. 75%酒精, 1次,1小时,
4. 85%酒精, 1次,1小时,
5. 95%酒精, 3次,1小时,
6. 100%酒精, 3次,1小时,
7. 二甲苯, 2次,1小时,
8. 石蜡浸泡, 3次,2小时,
HE 染色:
{水化}
1. 二甲苯, 2次,5-10分钟,
2. 100%酒精, 1次,1-2 分钟,
3. 95%酒精, 1次,1-2 分钟,
4. 85%酒精, 1次,1-2 分钟,
5. 75%酒精, 1次,1-2 分钟,
6. 过蒸馏水,
{染色}
1. Mayer 氏苏木素,1 分钟,
2. 温水冲洗, 5-10 分钟,
3. 75% 盐酸酒精, 1-2 分钟,
4. 自来水冲洗, 30 秒钟,
5. 依红复染, 1-2 分钟,
6. 自来水洗, 30 秒钟,
7. 85%酒精, 1次,1-2 分钟,
8. 95%酒精, 2次,1-2 分钟,
9. 100%酒精, 2次,1-2 分钟,
10. 二甲苯, 2次,5-10分钟,
11. 中性树胶封片。
免疫组化染色
一.石蜡切片免疫组化染色实验步骤:
1.石蜡切片脱蜡至水:(石蜡切片染色前应置60℃ 1小时)。
(1)二甲苯I、II,各10分钟。
(2)梯度酒精:100%,2分钟® 95%,2分钟® 80%,2分钟® 70%,2分钟。
(3)蒸馏水洗:5分钟,2次(置于摇床)。
2.过氧化氢封闭内源性过氧化物酶:3%H2O2,室温10分钟(避光)。
3.蒸馏水洗:5分钟,2次(置于摇床)。
4.抗原修复:根据待检测的抗原,选择适当的方法。
附:抗原修复液(10mM pH 6.0枸橼酸钠缓冲液)的配制
(1)储备液的配制:
A液:枸橼酸三钠-2H2O 29.41g + 蒸馏水1000ml
B液:枸橼酸 21g + 蒸馏水1000ml
(2)工作液的配制:A液82ml + B液18ml + 蒸馏水900ml
抗原修复的方法:
(1)高压锅处理技术:枸橼酸钠缓冲液(10mM,PH6.0),淹没切片,盖 上锅盖,高压锅内煮沸,上汽3分钟后缓慢冷却(可用自来水在高压锅外冲,以助冷却)。
(2)微波处理技术:用塑料切片架,置于塑料或耐温玻璃容器内,枸橼酸钠缓冲液淹没切片,选择中高或高档,5分钟;取出并补充已预热的枸橼酸钠缓冲液;再选择中高或高档,5分钟。(最佳温度为92~95℃)
(3)酶消化处理:此略。
抗原修复的注意事项:
(1)组织不能干。
(2)选择抗原修复方法要因抗体而异。
(3)该方法主要用于10%福尔马林固定、石蜡包埋组织。
(4)抗原修复后至DAB显色的过程中,均需用PBS缓冲液。
5.PBS:5分钟,2次(置于摇床)。
6.正常血清封闭:从染片缸中取出切片,擦净切片背面水分及切片正面组织
周围的水分(保持组织呈湿润状态),滴加正常山羊或兔血清(与第二抗 体 同源动物血清)处理,37℃,15分钟。
附:正常血清配制 (或按试剂盒规定的浓度配制)
按1:20比例,用PBS配制,每张切片需要量按50ml+5ml (10%抛洒量)计算。
7.滴加第一抗体:用滤纸吸去血清,不洗,直接滴加第一抗体,37℃ 2小时
(也可置于4℃冰箱过夜)。
8.PBS:5分钟,2次(置于摇床)。
9.滴加生物素化的二抗,37℃,40分钟。
10.PBS:5分钟,2次(置于摇床)。
11.滴加三抗 (SAB复合物),37℃,40分钟。
12.PBS:5分钟,2次(置于摇床)。
13.DAB 显色,镜下观察,适时终止(自来水冲终止)。
附:DAB的配制
(1)储备液(DAB 25mg/ml)的配制:DAB 250mg + PBS 10ml,待完全溶解后分装成 1ml,100ml,50ml,20ml等,—20℃,冻存。
(2)工作液:DAB 储备液20ml + PBS 1000ml + 3% H2O2 5ml
14.自来水(细水)充分冲洗。
15.苏木素复染,室温,30秒,自来水冲洗。
16.自来水冲洗返蓝,15分钟。
17.梯度酒精脱水:
80%,2分钟 ® 95%,2分钟 ® 100%,2次,5分钟。
18.二甲苯透明:
I,II(二甲苯)各5分钟
19.封片:加拿大树胶(或中性树胶)封片。
二.细胞爬片的免疫组化染色:
1. 取出细胞爬片,迅速置入冷丙酮固定20 ~30分钟。
2. 蒸馏水浸泡5分钟,2次。
3. 打孔液浸泡5分钟。
4. 蒸馏水浸泡5分钟,2次。
5. 后接前述实验步骤的第6步(正常血清封闭)。
注:第3、4步仅用于检测细胞内抗原,检测细胞膜抗原时不用。
三.冰冻切片的免疫组化染色:
1. 新鲜组织立即在恒冷冰冻切片机内切片(也可-80℃保存),厚度为5~6mm。
2. 载玻片可不打底,裱片后,立即用电吹风吹干。
3. 如不马上染色,可密封后-20℃保存。
4. 染色前用冷丙酮在4℃固定10-20分钟。
5. PBS洗2次,每次5分钟,(必要时应用0.1%柠檬酸钠+0.1%triton打孔)
6. 3% H2o2灭活内源性过氧化物酶,20分钟,避光;
7. 用PBS 洗2次,每次5分钟;
8. 接前面实验步骤第六步.
四 .切片的预处理:
1. 洗衣粉液浸泡30分钟,冲洗,晾干。
2. 洗液 (含强酸、高锰酸钾等)浸泡24小时,冲洗,晾干。
3. APES(1:50 丙酮溶液) 10-20秒(注意:不能用塑料容器及塑料切片架)。
4. 纯丙酮 I,约10秒。纯丙酮 II,约5秒
5. 晾干或烤箱内烤干,备用。
流式细胞仪常用的几种检测方法
一、测定用乙醇固定的DNA的含量
1、培养细胞的DNA含量的测定
制备单细胞悬液于200μl的PBS缓冲液中;
加入2ml预冷的70%乙醇,4℃保存;
附:细胞固定的一般步骤
1) 取单细胞悬液1~2×106个细胞于PBS(PH=7.2)缓冲液中;
2) 300g离心5分钟,弃上清,反复两次;
3) 重悬细胞于0.5ml PBS缓冲液中;
4) 将细胞悬液放置于2~3ml冷70%乙醇中,混匀,保存于4℃,至少30分钟。在4℃条件下可保存2~3周。
注意:
² 根据实验的要求,固定剂也可选用1~3%多聚甲醛;
² 将乙醇作为固定剂时,乙醇应预冷至0~4℃;
² 细胞在固定时,固定剂应缓慢滴入细胞悬液中,使固定剂的浓度缓慢增加,并不断震摇,以免细胞成团(特别是用乙醇固定时)。
² 300g离心5分钟,去上清,再重悬于400μl PBS中;
² 显微镜下观察,若有明显的黏附,须再用筛网过滤;
² 加入PI(含Rnase),避光孵育30分钟;
² 上机检测。
2、新鲜组织的DNA含量的测定
1) 用200mg湿重组织用机械法制成单细胞悬液;
2) 500g离心5分钟;
3) 弃上清,重悬于10ml染色-去污剂中;
4) 再过滤,用200目的筛网或70~80μm的筛网过滤;
5) 上机检测。
3、石蜡包埋组织切片的DNA含量的测定
1) 从石蜡包埋切取切片50 μm厚,2~3片,制成单细胞悬液;
2) 用PBS缓冲液洗涤,500g离心5分钟,弃上清;
3) 加入PI液1ml室温避光30分钟;
4) 调整细胞浓度为1×106/ml;
5) 上机检测。
.
二、细胞凋亡检测及相关分子检测
1、细胞DNA含量分布(由细胞DNA降解方式检测细胞凋亡)
² 收集已固定的单细胞悬液约5×105~1×106/ml;
² 离心除去固定液,3ml PBS重悬细胞;
² 1500rpm离心,5分钟 ,弃去PBS;
² 加PI染液1ml,室温避光20分钟;
² 调整细胞浓度5×105/ml;
² 上机检测。
.
2、磷脂酰丝氨酸外翻分析检测细胞凋亡
1) 常规制备单细胞悬液,用PBS洗两次。(若为全血要先溶血),取约5×106个细胞,1500rpm离心,弃上清,用400μl 1×Binding Buffer重悬;
2) 分成a、b、c、d、e五管,每管约1×106个细胞
a) 阴性对照,不加任何试剂;
b) 阳性对照,加2%多聚甲醛固定30分钟,加AnnexinV 5μl,室温10min,用1×Binding Buffer洗一次,弃上清,再加190μl 缓冲液和10μl PI避光15分钟;
c) 加10μl PI,避光孵育15分钟;
d) 加5μl AnnexinV液,室温避光孵育15min;
e) 加10μl PI和5μl AnnexinV液,室温避光孵育5min;
3) 每管各加400μl 1×Binding Buffer。
4) 上机检测。
注意 a. 操作动作要尽量轻柔,勿用力吹打细胞;
b. 操作时注意避光;
c. 反应完毕后尽快检测,最好在一小时内检测。
3、用单克隆抗体APO2.7检测细胞凋亡
1) 放置0.5~1×106个细胞到试管中;
2) 室温离心200g,6min;
3) 弃上清,加入100μl冷的(4℃)在PBSF溶液中含100µg/ml的digitonnin,轻轻地重悬细胞,在冰上孵育20min;
4) 加入2ml冷的(4℃)PBSF液,室温离心200g,6min;
5) 弃上清,加入20μl PE标记的Apo2.7 单克隆抗体和80μl PBSF液,用vortex轻轻震荡,室温避光孵育15分钟;
6) 加入2ml PBSF液,室温离心200g,6min;
7) 弃上清,加入1ml PBSF液重悬细胞;
8) 避光保存,直到流式细胞仪检测。
PBSF:含2.5% FCS(v/v)和0.01%NaN3(w/v)的PBS。
三、用流式细胞术进行DNA周期分析
1、方法:同DNA含量检测
2、注意:
单细胞浓度应约106/ml,以免影响检测的CV值和检测结果;
制备完成后的标本应用光学显微镜检查其质量(如细胞是否聚集或过多碎片),以保证得到足够的细胞含量;
醛类固定会影响PI与核酸的结合。
四、免疫荧光标记法
1、细胞膜上的免疫荧光检测法
间接标记法
1) 制备单细胞悬液;
2) 细胞计数,取出1×106个细胞于试管中;
3) 用台盼蓝染色计活细胞数,要求活细胞数 >90~95%;
4) 在试管中加入一抗,(剂量按照说明书的要求),孵育30~60分钟;
5) PBS洗涤1~2次,1500rpm,离心3分钟,弃上清;
6) 加入二抗,孵育20~30分钟;
7) PBS洗一次,1500rpm,离心3分钟,弃上清;
8) 加300μl PBS,上机检测(若不能及时上机检测,可加1ml 1%多聚甲醛固定,可放置1周)。
直接标记法
①~③同间接标记法
④在试管中加入荧光标记的抗体,混匀,孵育30分钟;
⑤用PBS洗2次,1500rpm,离心3分钟,弃上清;
⑥加300μl PBS(PH=7.4),上机检测。
2、 细胞膜内的免疫荧光标记法
间接免疫荧光标记法
1) 取已制备好的单细胞悬液,用1~3%的多聚甲醛固定30分钟(也可4℃保存过夜);
2) 用PBS洗两次,弃上清;
3) 细胞膜打孔,加入0.1%皂素 200μl,室温10分钟;
4) 用PBS洗涤两次;
5) 加入第一抗体,室温30~60分钟,或4℃过夜,同时须设阴性对照或同型对照管;
6) 用PBS洗涤两次;
7) 加入二抗(荧光标记的抗体)室温20分钟,避光;
8) 用PBS洗涤1~2次,弃上清;
9) 重悬细胞于500μlPBS中,上机检测。
直接荧光标记法
①~⑤同间接荧光标记法;
加入荧光素标记好的抗体,避光30分钟(同时做同型对照管);
用PBS洗涤1~2次,弃上清;
加300μl PBS上机检测。
细胞膜上及细胞内双标记法(直标法)
1) 取出已制备好的未固定的单细胞悬液1×10 6个细胞于试管中;
2) 用PBS洗涤两次;
3) 加入用荧光素标记的抗体来标记细胞膜上的抗原,同时加上阴性对照管,室温孵育20分钟;
4) 在试管中加入1%~3%多聚甲醛1ml,固定30分钟;
5) PBS洗涤两次1500rpm 3分钟,弃上清;
6) 打孔,加入0.1%皂素200μl室温10分钟;
7) 用PBS洗涤两次,弃上清;
8) 加入用荧光素标记的抗体,标记膜内的抗原(标记膜内的抗体的荧光素的颜色与膜上标记的荧光素的颜色务必不相同)室温20分钟孵育;
9) 用PBS洗一遍弃上清;
10) 用300μlPBS重悬细胞,上机检测
五、流式细胞术中的几点注意事项:
1、对照组的设置:
在流式细胞术中所测得的量都是相对值,不是绝对值。如需知道绝对值时必须设置对照组样品。对照组样品包括有阴性对照和阳性对照。
(1)、阴性对照的设置
² 在实验过程中,如做间接标记法,可设置与一抗无关的实验,即在实验中不加一抗而只加上带有荧光标记的第二抗体作为阴性对照管,作为阴性对照。
² 在实验过程中,假设做直接标记法,可设置理论上的阴性细胞作为阴性对照管,实验过程及步骤与实验组务必相同。(做间接标记法时,同样也可同时设置“阴性细胞”的阴性对照管作为阴性对照。
² 在实验过程中,假设做直接标记法,可将实验组细胞,取一管,加上与实验抗体所标记的荧光颜色相同的同型对照来作为阴性对照。
(2)、阳性对照的设置:
在实验过程中如涉及到表达缺失或减少的实验,应设置阳性对照组,其设置方法与阴性对照设置相同。
2、几点建议:
1) 在实验过程中,在保证实验的科学性和准确性的基础上,应尽量减少实验工序和过程。由于间接标记法的工序多,实验过程长,如再加之操作的不熟练,细胞更容易丢失和受损,而造成实验结果的误差。因此,在条件允许的范围内,建议尽量做直接标记法而不去做间接标记法,以保证实验的真实性和准确性。
2) 建议送检细胞一定要足够量,一般要求1×106个细胞。不要过少。因为,如细胞太少检测时样本流量相对会增大从而影响变异系数,结果也不可信。细胞量也不宜过多,因细胞量太多加入的抗体或染料相对不足,结果也由此受影响。
3) 同一种细胞需同时做双标记时,须做双标记的同型对照,且两种抗体所标记的荧光颜色务必不同。
Western Blot(免疫印迹法)
主要包括以下4个基本步骤:
n 样品制备
n 电泳分离
n 蛋白的膜转移
n 免疫杂交与显色――蛋白检测
溶液和试剂
n 1X 磷酸盐缓冲液(PBS)
n Modified RIPA buffer
Tris-HCl: 50 mM, pH 7.4 ; NP-40: 1% ;Na-deoxycholate: 0.25% ;NaCl: 150 mM ;EDTA: 1 mM ;PMSF: 1 mM ;Aprotinin, leupeptin, pepstatin: 1 microgram/ml each ;Na3VO4: 1 mM ;NaF: 1 mM
n 1X SDS 样品缓冲液
62.5 mM Tris-HCl (pH 6.8 于 25°C), 2% w/v SDS, 10%甘油,50 mM DTT, 0.01% w/v溴酚蓝
n 转移缓冲液
25 mM Tris base, 0.2 M 甘氨酸, 20%甲醇 (pH 8.3)
n 10X Tris缓冲盐 (TBS)
准备1L 10X TBS: 24.2 g Tris base, 80 g NaCl;用1N HCl调pH为 7.6
n 脱脂奶粉或BSA
n 甲醇
n TBS/T缓冲液
1X TBS, 0.1% Tween-20
n 封闭缓冲液(TBS/T)
1X TBS, 0.1% Tween-20加5% w/v脱脂奶粉或BSA
n 一抗的稀释
1X TBS, 0.1% Tween-20 加 5% BSA (多抗)或 5%脱脂奶粉(单抗)
Note: 一般来说, BSA被推荐用于多克隆抗体,脱脂奶粉用于单克隆抗体,这样可得到较高的信噪比。抗体的稀释度参考抗体说明书或根据实验确定。
n 预染的蛋白质Marker,可用于监测转膜的效率
样品制备
原始样品可为细胞、组织、培养上清、免疫沉淀或亲和纯化的蛋白,以下为定性检测目的蛋白时细胞样品的处理方法,其余的样品制备方法参阅相关文献。
1.培养细胞或药物处理。
2.弃培养基,用1X PBS漂洗细胞2次,去尽残留培养基。
3.加入1X SDS样品缓冲液(6-well plate, 100 µl /w或 75 cm2 plate, 500-1000 µl/瓶),刮落细胞,转移到Ep管。注意:冰上操作。
4.超声 10~15秒剪切DNA以减低样品粘性。
5.煮沸样品5 minutes。
6.离心12000g, 5 min,取上清。
7.电泳分离:上样15µl~20 µl 至 SDS-PAGE 胶 (10 cm x 10 cm)电泳。
如要定量检测某蛋白的表达水平,应用RIPA裂解液(1 ml per 107 cells/100 mm dish/150 cm2 flask)裂解细胞,收集裂解液至离心管中,在振荡器上混匀4~15min,14000g离心15min(4℃),弃沉淀,用Bradford法或其它蛋白质测定方法测定上清中蛋白浓度以调整上样体积和上样量,进行Western杂交时还需设置内或外参照,通常用beta-actin。
注意:一般上样20~30 µg已足够,如待检蛋白为低丰度蛋白,可加大上样量至100µg,但电泳条带易拖尾,可制备亚细胞组份或采用更敏感的检测方法。
电泳分离(参照SDS-PAGE电泳方法)
转膜
杂交膜的选择是决定Western blot成败的重要环节。应根据杂交方案、被转移蛋白的特性以及分子大小等因素,选择合适材质、孔径和规格的杂交膜。用于Western blot的膜主要有两种:硝酸纤维素膜(NC) 和PVDF膜。NC膜是蛋白印迹实验的标准固相支持物,在低离子转移缓冲液的环境下,大多数带负电荷的蛋白质会与膜发生疏水作用而高亲和力的结合在一起,但在非离子型的去污剂作用下,结合的蛋白还可以被洗脱下来。根据被转移的蛋白分子量大小,选择不同孔径的NC膜。因为随着膜孔径的不断减小,膜对低分子量蛋白的结合就越牢固。通常用0.45µm和0.2µm两种规格的NC膜。大于20kD的蛋白可用0.45µm的膜,小于20kD的蛋白就要用0.2µm的膜了,如用0.45µm的膜就会发生“Blowthrough”的现象。PVDF膜灵敏度、分辨率和蛋白亲和力比常规的膜要高,非常适合于低分子量蛋白的检测。但PVDF膜在使用之前必需用纯甲醇浸泡饱和1-5秒钟。
蛋白质常用的转移方法主要有两种:槽式湿转和半干转移。前者操作容易,转移效率高;而后者适用于大胶的蛋白转移,所用缓冲液少。以下为槽式湿转的操作步骤。
1. 将胶浸于转移缓冲液中平衡10min。
注意:如检测小分子蛋白,可省略此步,因小分子蛋白容易扩散出胶。
2. 依据胶的大小剪取膜和滤纸6片,放入转移缓冲液中平衡10min。如用PVDF膜需用纯甲醇浸泡饱和3-5秒钟。
3. 装配转移三明治:海绵®3层滤纸®胶®膜®3层滤纸®海绵,每层放好后,用试管赶去气泡。切记:胶放于负极面(黑色面)。
4. 将转移槽置于冰浴中,放入三明治(黑色面对黑色面),加转移缓冲液,插上电极,100V,1h(电流约为0.3A)。注意:应再次检查三明治和电极是否装配正确,电源是否接通。
5. 转膜结束后,切断电源,取出杂交膜

免疫杂交与显色
1.用25 ml TBS 洗膜5min,室温,摇动。
2.置膜于25 ml 封闭缓冲液中1h, 室温,摇动。
3.15ml TBS/T洗3次(5 min/T)。
4.加入合适稀释度的一抗,室温孵育1-2h或4°C过夜,缓慢摇动。
5.15 ml TBS/T洗3次(5 min/T)。
6.加入合适稀释度的碱性磷酸酶(AP)或辣根过氧化酶(HRP)标记的二抗,室温孵育1h,缓慢摇动。
7.15 ml TBS/T洗3次(5 min/T)。
8.15 ml TBS洗1次。
9.蛋白检测(显色法或发光法,按相应试剂说明操作)。
注意事项:
1.操作中戴手套,不要用手触膜。
2.PVDF膜在甲醇中浸泡时间不要超过5秒。
3.如检测小于20kD的蛋白应用0.2µm的膜,并可省略转移时的平衡步骤。
4.某些抗原和抗体可被Tween-20 洗脱,此时可用1.0% BSA代替Tween-20。
5.关于封闭剂的选择:5%脱脂奶/TBS or PBS: 能和某些抗原相互作用,掩盖抗体结合能力;0.3~3% BSA in PBS:低的内源性交叉反应性。
6.如用0. 1% Tween 20、0.02% NaN3 in PBS or TBS作封闭剂和抗体稀释液,抗体检测后可进行蛋白染色。
7.如要同时检测大分子量和小分子蛋白,最好用梯度胶分离蛋白。
PVDF膜上蛋白的可逆染色
Western杂交时,为确认蛋白是否转至PVDF膜上,可用下列方法对膜上蛋白进行可逆性染色。
1. 氨基黑染色
染液:0.5% Amido Black (w/v), 25% isopropanol (v/v) and 10% acetic acid.
染色:将PVDF膜置于染液中染色数钟,ddH2O 脱色。
2. 考马氏亮蓝染色
染液:0.1% Coomassie Brilliant Blue R-250 (w/v) and 50% methanol (v/v)
脱色液:40% methanol (v/v) with 10% acetic acid (v/v)
染色:将PVDF膜置于染液中染色15 min.,脱色液脱色。
3.丽春红染色Ponceau S
染液:0.2% w/v Ponceau S in TCA (3% v/v)
染色:将PVDF膜置于染液中染色5min
脱色:ddH2O 脱色
三、银染
PAGE胶上蛋白质的银染方法有数百种,其原理相似,具体步骤各不相同。银染为蛋白质的非特异性染色,呈“爆炸性”反应模式,用于蛋白定量时准确性差,但其敏感度高、简便易行,仍被广泛应用。下面列举的这三种方法为常用的双向电泳凝胶染色法,可与质谱兼容。其中方法1敏感性最高,方法3背景最低、对比度好。
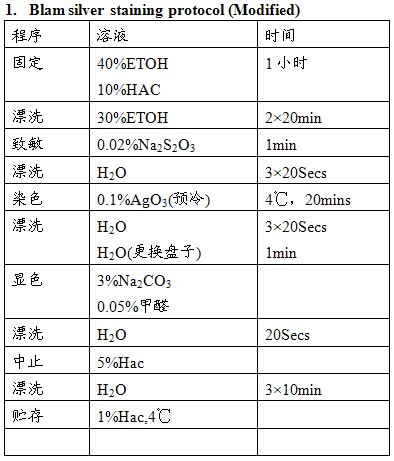

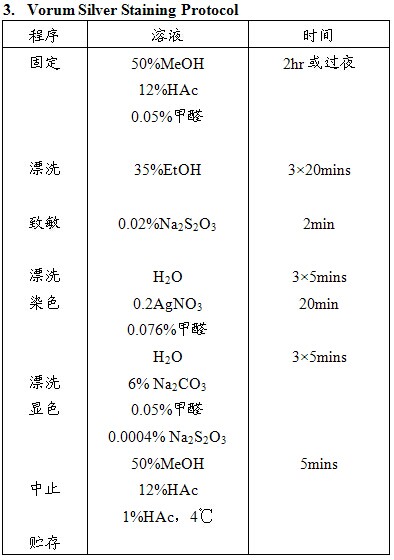
注意事项:
1. 应严格按照操作步骤进行;
2. 显色前最好更换新染色盘;
3. 显色时变为黄色或棕色后立即弃去,更换新鲜显色液,一般来说染一块胶应配制500ml染液。更换2-3次;
4. 市售甲醛的浓度为37%;
5. 三种方法均与质谱兼容,Blum法敏感性高,但背景呈淡黄色,EMBL法次之但背景较低,染色点较黑。Vorum相对不敏感,但背景清晰,信噪比高。
人肿瘤抗原的识别与鉴定
-免疫沉淀实验流程
一、裂解细胞
1.收集细胞。将培养瓶置于冰上,倒掉培养基,用PBS或生理盐水漂洗两次,留适量液体于瓶内,然后用特制的细胞刮棒朝一个方向刮取细胞。将细胞悬液转移到离心管,离心取沉淀,-800C保存。(收集细胞要迅速,低温下进行,以减弱蛋白酶的作用)
2.裂解细胞。采用RIPA裂解体系,使用前40C预冷,按107个细胞加入1.0ml裂解液,吹打混匀,加入适量的蛋白酶抑制剂(如cocktail),冰上放置3~5分钟。
3.超声处理裂解液。使用探针型超声进行多次适当频率的短促冲击,10~20秒/次,重复3次,中间间隔10~20秒,冰浴下进行。
4.15,000rpm,40C离心15min,吸取上清液于另一Ep管中,置冰上。上清液用于下一步的预处理。
二、裂解物的预处理
使用正常人的血清对裂解物进行预处理。
1、按1ml裂解物加2µl对照血清的比例加入正常人血清。
2、室温孵育2~3小时,缓慢摇动。
三、免疫复合物的纯化
1) 用Dynabeads proteinA进行免疫复合物的纯化。
2) 将Dynabeads proteinA 振荡混匀,吸取100µl磁珠于Ep管中,置于磁铁架(Dynal MPC)上,磁珠吸附到管壁上,弃上清。
3) 加500µl 0.1M Na-phosphate (pH 8.1 )至Ep管,轻柔搓动管子约2~3min,将Ep管置于磁铁架上,磁珠吸附到管壁上,弃上清。
4) 重复2步骤两次。
5) 清洗完磁珠后,加500µl预处理后的裂解物,反复摇动管子,室温下反应10~20min。
6) 将Ep管置于磁铁架上,磁珠吸附到管壁上,将上清转移至另一Ep管中。
7) 用RIPA裂解液清洗磁珠3次。
8) 洗脱抗原-抗体复合物。加0.1M citrate (pH3.1) 30µl于Ep管中,轻柔搓动管子约2~3min,将Ep管置于磁铁架上,吸取上清于一Ep管。
9) 重复步骤7,将上清收集于同一个Ep管,洗脱样品总体积为60µl,作对照用。
10) 磁珠用 0.1M Na-phosphate (pH 8.1 ) 清洗三次后备用。
11) 在步骤5留取的上清中加入病人血清(1ml加2µl血清),缓慢摇动,室温孵育2~3h。
12) 将Ep管置于磁铁架上,磁珠吸附到管壁上,弃上清。
13) 重复步骤6~8。共收集到两份样品,对照与病人的纯化免疫复合物各60µl。
14) 磁珠用 0.1M Na-phosphate (pH 8.1 ) 清洗三次后,加入柱储液100µl,40C保存。
15) 在样品中加入2×SDS上样缓冲液,并用1M NaOH调节pH至使溴酚蓝呈蓝色。
四、1D SDS-PAGE
用免疫沉淀后的样品可直接进行一维凝胶电泳,或者对磁珠上的蛋白A或蛋白G进行耦联剂修饰,洗脱后的样品可进行Western Blot。
RIPA裂解液:150mmol/L NaCl;1.0%NP-40;0.5%脱氧胆酸钠;0.1%SDS;50mmol/L Tris,(pH 8.0)
蛋白质组实验流程
 |
双向电泳的样品的制备
样品制备原则
样品制备是双向电泳中最关键的一步,将直接影响2-DE结果好坏。目前并没有一个通用的样品制备方法,尽管处理方法多种多样,但都遵循几个基本的原则:1)尽可能的提高样品蛋白的溶解度,抽提最大量的总蛋白,减少蛋白质的损失;2)减少对蛋白质的人为修饰;3)破坏蛋白质与其他生物大分子的相互作用,并使蛋白质处于完全变性状态。
根据这一原则,样品制备需要四种主要的试剂:离液剂(chaotropes),主要包括尿素(Urea)和硫脲(thiourea);表面活性剂(sufactants),也称去垢剂,如CHAPS与Zwittergent系列等双性离子去垢剂;还原剂(reducing agents),最常用的是二硫苏糖醇(DTT)和磷酸三丁酯(TBP)等。当然,也可以选择性的加入Tris-base,蛋白酶抑制剂以及核酸酶。
样品的来源不同,其裂解的缓冲液也各不相同。通过不同试剂的合理组合,以达到对样品蛋白的最大抽提。在对样品蛋白质提取的过程中,必须考虑到去除影响蛋白质可溶性和2DE重复性的物质,比如核酸、脂、多糖等大分子以及盐类小分子。大分子的存在会阻塞凝胶孔径,盐浓度过高会降低等电聚焦的电压,甚至会损坏IPG胶条,这样都会造成2-DE的失败。样品制备的失败很难通过后续工作的完善或改进获得补偿。
核酸的去除可采用超声或核酸酶处理,超声处理应控制好条件,并防止产生泡沫;而加入的外源核酸酶则会出现在最终的2D胶上。脂类和多糖都可以通过超速离心除去。透析可以降低盐浓度,但时间太长;也可以采取凝胶过滤或沉淀/重悬法脱盐,但会造成蛋白质的部分损失。
因此,样品制备方法必须根据不同的样品、所处的状态以及实验目的和要求来进行选择。目前有很多方法适于2-DE,如组织或细胞的总蛋白提取物、亚细胞组份或细胞器蛋白、免疫沉淀的蛋白及其它亚组份蛋白(如磷酸化蛋白、采用亲合纯化凝集素结合蛋白等)。
一、细胞样品
1. 细胞培养,加药与处理。
2. 胰酶消化贴壁细胞,PBS漂洗3次(1500g,5min),弃上清, 再次离心,去尽残液(非常重要!)。如要比较细胞膜蛋白组的差别,最好用细胞刮收获细胞。如用10mM Tris/ 250 mM Sucrose(pH 7.0)代替PBS,可有效降低样品的盐浓度。
加入5倍体积裂解液,混匀(或将1×106细胞悬于60~100µl裂解液中)。
3. 加50ug/ml RNase及200ug/ml Dnase,在4℃放置15分钟。
4. 15,000转,4℃离心60分钟(或40,000转,4℃离心30分钟)。
5. 收集上清。
6. 测定蛋白浓度(采用BioRad RC/DC protein assay kit)。
7. 分装样品,冻存于-70℃。
二、组织样品
1. 碾钵碾磨组织,碾至粉末状。
2. 将适量粉末状组织转移至匀浆器,加入适量裂解液,进行匀浆。
3. 加50ug/ml RNase及200ug/ml DNase,在4℃放置15分钟。
4. 15,000转,4℃离心60分钟(或40,000转,4℃离心30分钟)。
5. 收集上清,测定蛋白浓度。
6. 分装样品,冻存于-70℃。
注意事项:
1. 8 mmol/L PMSF必须在添加还原剂之前用,否則PMSF会失去活性。
2. 40 mmol/L浓度以下的Tris可使有些蛋白酶在高pH值下失活。
3. 细胞清洗――大多用PBS,若PBS残留于细胞表面会造成胶上出现水平条纹,则可利用(10 mmol/L Tris, 250 mmol/L sucrose pH 7.0)來解決此问题。
双向电泳操作步骤
水化上样(被动上样)
1. 从冰箱中取出IPG胶条,室温放置10min。
2. 沿水化盘槽的边缘从左向右线性加入样品,槽两端各1cm左右不加样,中间的样品液一定要连贯。注意:不要产生气泡,否则会影响胶条中蛋白质的分布。
3. 用镊子轻轻撕去IPG胶条上的保护层。注意:碱性端较脆弱,应小心操作。
4. 将IPG胶条胶面朝下轻轻置于水化盘中样品溶液上。注意:不要将样品溶液弄到胶条背面,因为这些溶液不会被胶条吸收;还使胶条下面的溶液产生气泡。如产生了气泡,用镊子轻轻地提起胶条的一端,上下移动胶条,直到气泡被赶走。
5. 放置30~45min大部分样品被胶条吸收,沿着胶条缓慢加入矿物油,每根胶条约3ml(17cm IPG),防止胶条水化过程中液体蒸发。
6. 置等电聚焦仪于-20℃水化11~15h。
第一向 等电聚焦
1. 将纸电极置于聚焦盘的正负极上,加ddH2O 5~8µl润湿。
2. 取出水化好的胶条,提起一端将矿物油沥干,胶面朝下,将其置于刚好润湿的滤纸片上杂交以去除表面上的不溶物。
3. 将IPG胶条胶面朝下置于聚焦盘中,胶条的正极(标有+)对应于聚焦盘的正极,确保胶条与电极紧密接触。
4. 在每根胶条上覆盖2-3ml矿物油。
5. 对好正、负极,盖上盖子。设置等电聚焦程序。
6. 聚焦结束的胶条,立即进行平衡、第二向SDS-PAGE电泳。或将胶条置于样品水化盘中,-20℃冰箱保存,电泳前取出胶条,室温放置10分钟,使其溶解。
第二向SDS-PAGE电泳
1. 配制12%的丙烯酰胺凝胶。
2. 待凝胶凝固后,倒去分离胶表面的MilliQ水、乙醇或水饱和正丁醇,用MilliQ水冲洗。
3. 配制胶条平衡缓冲液I
4. 在桌上先放置干的厚滤纸,聚焦好的胶条胶面朝上放在干的厚滤纸上。将另一份厚滤纸用MilliQ水浸湿,挤去多余水分,然后直接置于胶条上,轻轻吸干胶条上的矿物油及多余样品,这样可以减少凝胶染色时出现的纵条纹。
5. 将胶条转移至样品水化盘中,加入6ml(17cm IPG)平衡缓冲液I,在水平摇床上缓慢摇晃15分钟。
6. 配制胶条平衡缓冲液II。
7. 第一次平衡结束后,取出胶条将之竖在滤纸上沥去多余的液体,放入平衡缓冲液II中,继续在水平摇床上缓慢摇晃15分钟。
8. 用滤纸吸去SDS-PAGE胶上方玻璃板间多余的液体,将二向凝胶放在桌面上,凝胶的顶部面对自己。
9. 将琼脂糖封胶液加热溶解。
10. 在100ml量筒中加入TGS电泳缓冲液。
11. 第二次平衡结束后,取出胶条,用滤纸吸去多余的平衡液(将胶条竖在滤纸上,以免损失蛋白或损坏凝胶表面)。
12. 用镊子夹住胶条的一端使胶面完全浸末在1×电泳缓冲液中漂洗数次。
13. 将胶条背面朝向玻璃板,轻轻放在长玻板上,加入低熔点琼脂糖封胶液。
14. 用适当厚度的胶片,轻轻地将胶条向下推,使之与聚丙烯酰胺凝胶胶面完全接触。注意:不要在胶条下方产生气泡,应推动凝胶背面的支撑膜,不要碰到胶面。
15. 放置5分钟,使低熔点琼脂糖封胶液凝固。
16. 打开二向电泳制冷仪,调温度为15℃。
17. 将凝胶转移至电泳槽中,加入电泳缓冲液,接通电源,起始时用的低电流(5mA~10mA/gel/17cm),待样品在完全走出IPG胶条,浓缩成一条线后,再加大电流(20-30mA/gel/17cm),待溴酚蓝指示剂达到底部边缘时即可停止电泳。
18. 电泳结束后,轻轻撬开两层玻璃,取出凝胶,并切角以作记号(戴手套,防止污染胶面)。
19. 进行染色。

二、考马斯亮蓝染色
CBB染色液
0.5%考马斯亮蓝G250或R250
40%甲醇
10%乙酸
将CBB溶于甲醇中并不停的搅拌15min。加入乙酸与ddH2O
脱色液:30%甲醇
10%乙酸
染色方法:1.在摇床上染色30min.
2.脱色至蛋白点或条带清晰可见
3. ddH2O洗3-5次
三、胶体考马氏亮蓝染色Colloidal Coomassie Staining(Cambridge centre for porteomics)
sensitivity = ~100ng
1. 固定:甲醇/醋酸/H2O(45:1:54)至少20min.
2. 染色12-18hr。
染色液: 17% (w/v) 硫酸铵
34%甲醇
0.5%醋酸
0.1% (w/v) Coomassie G250
3. 脱色:用H2O脱色至蛋白点和背景清晰.
双向电泳蛋白点的切取和保存
1. 用PDQuest软件或肉眼比对,找出感兴趣的蛋白点,并做好标记和记录。
2. 用MilliQ水冲洗胶2次。
3. 用色谱纯甲醇和MilliQ水冲洗Ep管
4. 将枪头(200µl)下端剪去,使其内径略小于蛋白斑点的直径,用色谱纯甲醇和MilliQ水冲洗枪头。
5. 对准斑点中央小心将蛋白切割下来,放入Ep管,MilliQ水漂洗2次,如胶块太大,将其切成1 x 1 mm的胶片。
6. 将切好的点做好标记和记录,置-80oC保存或冻干后-20oC存放。
注意事项:
1. 尽量避免皮肤和头发的角蛋白的污染,在操作过程中应戴一次性的PE手套(不用乳胶手套)和帽子。
2. 不要将胶长期存放于乙酸溶液中。
3. Ep管及染胶的容器必须用甲醇和水充分清洗,尤其应与进行Western blotting的容器分开,以避免casein或BSA的污染。
数据库搜索(Databases search)
一般来说,利用质谱数据鉴定蛋白质主要有两种方法:肽质量指纹图谱(PMF)和肽序列标签分析(sequence tag analysis)。这两种方法较传统的N端测序或内部Edman测序法敏感数百倍,其检测低限为飞摩尔(fmol)水平(如银染的2D胶点或1D胶带)。然而,足够多的样品蛋白量可以增加识别的成功率,这是因为增大样品蛋白量可以克服一些污染物的干扰(如角蛋白,在样品中的存在非常常见)。数据库的检索是利用计算机程序算法将实验测得的肽质量数据与蛋白质序列数据库中的蛋白质的肽质量计算值进行相关性比较分析,从而得出可能蛋白质的概率并将相关性最好的结果排序,这是凝胶分离蛋白质鉴定的最常用手段。
这种检索途径有一个明显的限制,就是被鉴定的蛋白质必须在序列数据库(蛋白质数据库和核酸序列翻译数据库)中存在。PMF检索鉴定蛋白质是依据实验中获得的蛋白质的多个肽段质量数据与同一蛋白质肽段质量理论计算值的之间的相关性比较。因此,这一技术不适用于检索EST翻译序列,也不适用于鉴定蛋白质的混合物。而肽序列标签分析(sequence tag analysis)可适用于检索EST数据库。
对PMF 数据,主要搜索Swiss Prot (速度快但不全面)和NCBI (速度虽慢但包含更多的信息)数据库。如果你认为你的蛋白质可能为新的蛋白质,选择NCBI数据库,标准搜索选择Swiss Prot 数据为好。如果需要,也可搜索EST数据库(用MS/MS数据按搜索更有效)。
检索条件的设置与结果判断:
1. 肽片段质量选择在800-4000Da范围。
2. 氨基酸残基的修饰(Modifications):半胱氨酸为脲甲基半胱氨酸(Cardamidomethyl-Cys),蛋氨酸选择可变修饰—氧化。
3. 最大允许的肽质量误差(Mass Tolerances),与仪器性能和数据质量有关,一般设为±0.1Da,最大为±0.5Da,质量误差愈小,搜索的特异性愈高。
4. 不完全酶解位点数目(Missed cleavages):每个肽允许有2个不完全裂解位点,一般选1(如果蛋白充分变性且酶解完全)。
5. 参考蛋白质表观分子量和等电点,表观PI误差范围为±0.5pH,表观Mr误差为范围为±20%。一般情况下不选此项,在判读结果时参考。
6. 物种选择:限定物种。
7. 离子选择:[M+H]+,单同位素
8. 最少匹配肽片段规定为4。
主要的公共数据库及网址
Mascot
http://www.matrixscience.com/home.html
MOWSE
http://www.seqnet.dl.ac.uk/Bioinformatics/Webapp/mowse/
Peptide Search
http://www.mann.embl-heidelberg.de/Services/PeptideSearch/
Protein Prospector
http://prospector.ucsf.edu/
Prowl
http://prowl.rockefeller.edu/程序:ProFound(www.expacy.ch)
EXPASY中国镜像站点(BI peptident)
双向电泳常见问题与原因
IEF
|
| Too much salt in the sample (disturbs IEF)
acetone precipitation to remove salts and other contaminants |
|
| Charged impurities in the sample acetone precipitation to remove salts and other contaminants |
|
| Impurities in the sample or rehydration solution acetone precipitation to remove salts and other contaminants Prepare new rehydration solution
|
|
| Lower pH left (e.g. pH 4-7): overfocused Prolong or lower the focusing time of the IEF |

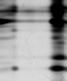

SDS-PAGE, staining
|
| Gel surface during polymerization not overlaid with water (or too low amount of water used) Apply at least 1 ml to overlay the gel surface | ||
|
| No uniform gel polymerization (e.g. impurities at gel cassettes, air bubbles in the polymerized gel) Clean gel cassettes with ethanol Cast the slab gel slowly (approx. 1 min) Overlay the gel carefully with destilled water | ||
|
| Not all proteins (especially high molecular mass proteins) saturated with SDS
Use 0.15 % instead of 0.1 % (w/ v) SDS in the 1 x SDS buffer for SDS-PAGE
| ||
| High sample load (protein disturbs of other spots or may not be fully saturated with SDS) Lower the protein amount | ||
|
|
Impurities on or within the 2-D gel still present during silver staining Clean the gel cassettes prior casting with ethanol Incubate the 2-D gels long enough (and with at least 100 ml/ gel) in fixing and washing solution prior staining |

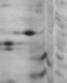



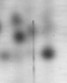
蛋白质的序列分析流程
1 蛋白质序列的检索
1.1 从NCBI检索蛋白质序列
http://www.ncbi.nlm.nih.gov:80/entrez/query.fcgi?db=Protein
1.2 利用SRS系统从EMBL检索蛋白质序列
http://srs.ebi.ac.uk/
2 蛋白质序列的基本性质分析
2.1 蛋白质序列的信号肽分析
http://genome.cbs.dtu.dk/services/SignalP-2.0/
http://genome.cbs.dtu.dk/services/SignalP/
2.2 蛋白质序列的跨膜区分析
http://genome.cbs.dtu.dk/services/TMHMM-1.0/
http://www.ch.embnet.org/software/TMPRED_form.html
2.3 蛋白质序列的亚细胞定位分析
http://predict.sanger.ac.uk/nnpsl/nnpsl_mult.cgi
3 蛋白质序列的同源性分析
3.1 基于NCBI/Blast软件的蛋白质序列同源性分析
http://www.ncbi.nlm.nih.gov/blast/blast.cgi
3.2 基于WU/Blast2软件的蛋白质序列同源性分析
http://dove.embl-heidelberg.de/Blast2/
3.3 基于FASTA软件进行蛋白质序列同源性分析
http://www2.ebi.ac.uk/fasta3
3.4 两条蛋白质序列之间的同源性分析
http://www.ncbi.nlm.nih.gov/gorf/bl2.html
3.5 蛋白质序列的批量联网同源性分析
4 蛋白质序列的结构功能域分析
4.1 蛋白序列的motif和Prosite分析
http://www.isrec.isb-sib.ch/software/PFSCAN_form.html
4.2 蛋白质的结构功能域分析
http://smart.embl-heidelberg.de/
http://www.ebi.ac.uk/interpro/interproscan/ipsearch.html
5 蛋白质家族分析及其进化树的构建(方案)
WEB RESOURCES FOR PROTEIN SCIENTISTS
http://www.faseb.org/protein/docs/WWWResources.html
蛋白质数据库(Protein databank, PD)由美国自然科学基金会、能源部和国立卫生研究院共同投资建立,主要由X-射线晶体衍射和核磁共振(NMR)测得的生物大分子三维结构所组成,用户可直接查询、调用和观察库中所收录的任何大分子三维结构。该数据库同时提供蛋白质序列及其三维空间晶体学原子坐标.其中受体-配体、抗原-抗体、底物-酶复合物等相互作用分子的共结晶图谱是基于同源比较的分子设计所需的最佳模型,因此PDB数据库为初步的蛋白质合理设计提供了重要的知识来源。由于PDB主要由生物大分子三维结构所组成,它具有以下几种功能:
(1)能够查找目的蛋白质的结构;
(2)可进行一级或高级结构的简单分析;
(3)与互联网上的其它一些数据库,如GDB、GenBank、SWISS-PROT、PIR等链接,从而可查询蛋白质的其它信息;
(4)可下载有关的结构信息以供进一步使用。可通过关键词或PDB 标识符等进行查询。
在序列分析中,PDB主要可应用于蛋白质结构预测和结构同源性比较。其中NRL-3D数据库则是PDB数据库中所有蛋白质序列的信息。该数据库允许进行基于结构的序列比较,网址为:http://www.rcsb.org/pdb/。


附录:
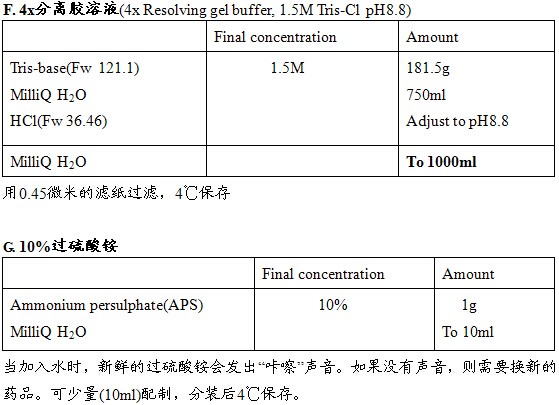
分子生物学常用溶液配制
一、分子生物学常用贮存液的配制
1.30%丙烯酰胺溶液
【配制方法】
将29g丙烯酰胺和1g N,N’-亚甲双丙烯酰胺溶于总体积为60ml的水中。加热至37℃溶解之,补加水至终体积为100ml。用滤器(0.45μm孔径)过滤除菌,查证该溶液的pH值应不大于7.0,置棕色瓶中保存于室温。
【注意】
丙烯酰胺具有很强的神经毒性并可以通过皮肤吸收,其作用具累积性。称量丙烯酰胺和亚甲双丙烯酰胺时应戴手套和面具。可认为聚丙烯酰胺无毒,但也应谨慎操作,因为它还可能会含有少量未聚合材料。
一些价格较低的丙烯酰胺和双丙烯酰胺通常含有一些金属离子,在丙烯酰胺贮存液中加入大约0.2体积的单床混合树脂(MB-1Mallinckrodt),搅拌过夜,然后用Whatman 1号滤纸过滤以纯化之。
在贮存期间,丙烯酰胺和双丙烯酰胺会缓慢转化成丙烯酰和双丙烯酸。
2.40%丙烯酰胺
【配制方法】
把380g丙烯酰胺(DNA测序级)和20g N,N’-亚甲双丙烯酰胺溶于总体积为600ml的蒸馏水中。继续按上述配制30%丙烯酰胺溶液的方法处理,但加热溶解后应以蒸馏水补足至终体积为1L。
【注意】
见上述配制30%丙烯酰胺的说明,40%丙烯酰胺溶液用于DNA序列测定。
3.放线菌素D溶液
【配制方法】
把20mg放线菌素D溶解于4ml 100%乙醇中,1:10稀释贮存液,用100%乙醇作空白对照读取OD440值。放线菌素D(分子量为1255)纯品在水溶液中的摩尔消化系数为21,900,故而1mg/ml的放线菌素D溶液在440nm处的吸光值为0.182,放线菌素D的贮存液应放在包有箔片的试管中,保存于-20℃。
【注意】
放线菌素D是致畸剂和致癌剂,配制该溶液时必须戴手套并在通风橱内操作,而不能在开放在实验桌面上进行,谨防吸入药粉或让其接触到眼睛或皮肤。
药厂提供的作治疗用途的放线菌素D制品常含有糖或盐等添加剂。只要通过测量贮存液在440nm波长处的光吸收确定放线菌素D的浓度,这类制品便可用于抑制自身引导作用。
4.0.1mol/L腺苷三磷酸(ATP)溶液
【配制方法】
在0.8ml水中溶解60mg ATP,用0.1mol/L NaOH调至pH值至7.0,用蒸馏水定容1ml,分装成小份保存于-70℃
5.10mol/L乙酸酰溶液
【配制方法】
把770g乙酸酰溶解于800ml水中,加水定容至1L后过滤除菌。
6.10%过硫酸铵溶液
【配制方法】
把1g过硫酸铵溶解于终量为10ml的水溶液中,该溶液可在4℃保存数周。
7.BCIP溶液
【配制方法】
把0.5g的5-溴-4-氯-3-吲哚磷酸二钠盐(BCIP)溶解于10ml 100%的二甲基甲酰胺中,保存于4℃
8.2×BES缓冲盐溶液
【配制方法】
用总体积90ml的蒸馏水溶解1.07g盐溶液BES[N,N-双(2-羟乙基)-2-氨基乙磺酸]、1.6g NaCl和0.027g Na2HPO4,室温下用HCl调节该溶液的pH值至6.96、然后加入蒸馏水定容至100ml,用0.22μm滤器过滤除菌,分装成小份,保存于-20℃。
9.1mol/L CaCl2溶液
【配制方法】
在200ml蒸馏水中溶解54g CaCl2·6H2O,用0.22μm滤器过滤除菌,分装成10ml小份贮存于-20℃。
【注意】
制备感受态细胞时,取出一小份解冻并用蒸馏水稀释至100ml,用Nalgene滤器(0.45μm孔径)过滤除菌,然后骤冷至0℃。
10.2.5mol/L CaCl2溶液
【配制方法】
在20ml蒸馏水中溶解13.5g CaCl2·6H2O,用0.22μm滤器过滤除菌,分装成1ml小份贮存于-20℃。
11.1mol/L二硫苏糖醇(DTT)溶液
【配制方法】
用20ml 0.01mol/L乙酸钠溶液(pH5.2)溶解3.09g DTT,过滤除菌后分装成1ml小份贮存于-20℃。
【注意】
DTT或含有DTT的溶液不能进行高压处理。
12.脱氧核苷三磷酸(dNTP)溶液
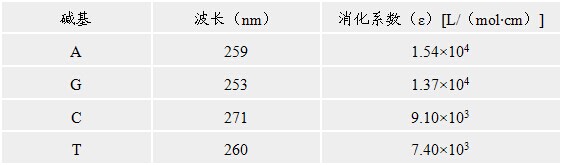
【配制方法】
把每一种dNTP溶解于水至浓度各为100mmol/L左右,用微量移液器吸取0.05mol/l Tris碱分别调节每一dNTP溶液的pH值7.0(用pH试纸检测),把中和后的每种dNTP溶液各取一份作适当稀释,在下表中给出的波长下读取光密度计算出每种dNTP的实际浓度,然后用水稀释成终浓度为50mmol/L的dNTP,分装成小份贮存于-70℃。
比色杯光径为1cm时,吸光度=εM
13.0.5mol/l EDTA(pH8.0)溶液
【配制方法】
在800ml水中加入186.1g二水乙二胺四乙酸二钠(EDTA-Na·2H2O),在磁力搅拌器上剧烈搅拌,用NaOH调节溶液的pH值至8.0(约需20g NaOH颗粒)然后定容至1L,分装后高压灭菌备用。
【注意】
EDTA二钠盐需加入NaOH将溶液的pH值调至接近8.0,才能完全溶解。
14.溴化乙锭(10mg/ml溶液)
【配制方法】
在100ml水中加入1g溴化乙锭,磁力搅拌数小时以确保其完全溶解,然后用铝箔包裹容器或转移至棕色瓶中,保存于室温。
【注意】
小心:溴化乙锭是强诱变剂并有中度毒性,使用含有这种染料的溶液时务必戴上手套,称量染料时要戴面罩。
15.2×HEPES缓冲盐溶液
【配制方法】
用总量为90ml的蒸馏水溶解1.6g NaCl、0.074g KCl、0.027g Na2PO4·2H2O、0.2g葡聚糖和1gHEPES,用0.5mol/l NaOH调节pH值至7.05,再用蒸馏水定容至100ml。用0.22μm滤器过滤除菌,分装成5ml小份,贮存于-20℃。
16.IPTG溶液
【配制方法】
IPTG为异丙基硫代-β-D-半乳糖苷(分子量为238.3),在8ml蒸馏水中溶解2g IPTG后,用蒸馏水定容至10ml,用0.22μm滤器过滤除菌,分装成1ml小份贮存于-20℃。
17.1mol/L乙酸镁溶液
【配制方法】
在800ml水中溶解214.46g四水乙酸镁,用水定容至1L过滤除菌。
18.1mol/L MgCl2溶液
【配制方法】
在800ml水中溶解203.4g MgCl2·6H2O,用水定容至1L,分装成小份并高压灭菌备用。
【注意】
MgCl2极易潮解,应选购小瓶(如100g)试剂,启用新瓶后勿长期存放。
19.β-巯基乙醇(BME)溶液
【配制方法】
一般得到的是14.4mol/L溶液,应装在棕色瓶中保存于4℃。
【注意】
BME或含有BME的溶液不能高压处理。
20.NBT溶液
【配制方法】
把0.5g氯化氮蓝四唑溶解于10ml 70%的二甲基甲酰胺中,保存于4℃。
21.酚/氯仿溶液
【配制方法】
把酚和氯仿等体积混合后用0.1mol/L Tris·HCl(pH7.6)抽提几次以平衡这一混合物,置棕色玻璃瓶中,上面覆盖等体积的0.01mol/l Tris·HCl(pH=7.6)液层,保存于4℃。
【注意】
酚腐蚀性很强,并可引起严重灼伤,操作时应戴手套及防护镜,穿防护服。所有操作均应在化学通风橱中进行。与酚接触过的部位皮肤应用大量的水清洗,并用肥皂和水洗涤,忌用乙醇。
22.10mmol/L苯甲基磺酰氟(PMSF)溶液
【配制方法】
用异丙醇溶解PMSF成1.74mg/ml(10mmol/L),分装成小份贮存于-20℃。如有必要可配成浓度高达17.4mg/ml的贮存液(100mmol/L)。
【注意】
PMSF严重损害呼吸道粘膜、眼睛及皮肤,吸入、吞进或通过皮肤吸收后有致命危险。一旦眼睛或皮肤接触了PMSF,应立即用大量水冲洗之。凡被PMSF污染的衣物应予丢弃。
PMSF在水溶液中不稳定。应在使用前从贮存液中现用现加于裂解缓冲液中。PMSF在水溶液中的活性丧失速率随pH值的升高而加快,且25℃的失活速率高于4℃。pH值为8.0时,20μmmol/l PMSF水溶液的半寿期大约为85min,这表明将PMSF溶液调节为碱性(pH>8.6)并在室温放置数小时后,可安全地予以丢弃。
23.磷酸盐缓冲溶液(PBS)溶液
【配制方法】
在800ml蒸馏水中溶解8g NaCl、0.2g KCl、1.44g Na2HPO4和0.24g KH2PO4,用HCl调节溶液的pH值至7.4加水定容至1L,在15lbf/in2(1034×105Pa)高压下蒸气灭菌20min。保存于室温。
24.1mol/L乙酸钾(pH=7.5)溶液
【配制方法】
将9.82g乙酸钾溶解于90ml纯水中,用2mol/L乙酸调节pH值至7.5后加入纯水定容到1L,保存于-20℃。
25.乙酸钾溶液(用于碱裂解)
【配制方法】
在60ml 5mol/L乙酸钾溶液中加入11.5ml冰乙酸和28.5ml水,即成钾浓度为3mol/L而乙酸根浓度为5mol/L的溶液。
26.3mol/L乙酸钠(pH5.2和pH7.0)溶液
【配制方法】
在80ml水中溶解408.1g三水乙酸钠,用冰乙酸调节pH值至5.2或用稀乙酸调节pH值至7.0,加水定容到1L,分装后高压灭菌。
27.5mol/L NaCl溶液
【配制方法】
在800ml水中溶解292.2g NaCl加水定容至1L,分装后高压灭菌。
28.10%十二烷基硫酸钠(SDS)溶液
【配制方法】
在900ml水中溶解100g电泳级SDS,加热至68℃助溶,加入几滴浓盐酸调节溶液的pH值至7.2,加水定容至1L,分装备用。
【注意】
SDS的微细晶粒易扩散,因此称量时要戴面罩,称量完毕后要清除残留在称量工作区和天平上的SDS,10%SDS溶液无须灭菌。
29.20×SSC溶液
【配制方法】
在800ml水中溶解175.3g NaCl和88.2g柠檬酸钠,加入数滴10mol/l NaOH溶液调节pH值至7.0,加水定容至1L,分装后高压灭菌。
30.20×SSPE溶液
【配制方法】
在800ml水中溶解17.5g NaCl、27.6g NaH2PO4·H2O和7.4g EDTA,用NaOH溶液调节pH值至7.4(约需6.5ml 10ml/L NaOH),加水定容至1L,分装后高压灭菌。
31.100%三氯乙酸溶液
【配制方法】
在装有500g TCA的瓶中加入227ml水,形成的溶液含有100%(M/V)TCA。
32.1mol/L Tris溶液
【配制方法】
在800ml水中溶解121.91g Tris碱,加入浓HCl调节pH值至所需值。
pHHCl
7.4 70ml
7.6 60ml
8.0 42ml
应使溶液冷至室温后方可最后调定pH值,加水定容至1L,分装后高压灭菌。
【注意】
如1mol/L溶液呈现黄色,应予丢弃并置备质量更好的Tris。
尽管多种类型的电极均不能准确测量Tris溶液的pH值,但仍可向大多数厂商购得合适的电极。
Tris溶液的pH值因温度而异,温度每升高1℃,pH值大约降低0.03个单位。例如:0.05mol/L的溶液在5℃、25℃、和37℃时的pH值分别为9.5、8.9和8.6。
33.Tris缓冲盐溶液(TBS)(25mmol/l Tris)
【配制方法】
在800ml蒸馏水中溶解8g NaCl、0.2g KCl和3g Tris碱,加入0.015g酚并用HCl调至pH值至7.4,用蒸馏水定容至1L,分装后在151bf/in2(1.034×105Pa)高压下蒸汽灭菌20min,于室温保存。
34.X-gal溶液
【配制方法】
X-gal为5-溴-4-氯-3-吲哚-β-D半乳糖苷。用二甲基甲酰胺溶解X-gal配制成的20mg/ml的贮存液。保存于一玻璃管或聚丙烯管中,装有X-gal溶液的试管须用铝箔封裹以防因受光照而被破坏,并应贮存于-20℃。X-gal溶液无须过滤除菌。